一種藥物及其制備方法和應用與流程
本發明屬于腫瘤免疫治療領域,涉及一種藥物及其制備方法和應用,具體涉及一種具有提高抗原加工功能的藥物及其制備方法和應用。
背景技術:
近年來,腫瘤免疫治療因為對于常規無效的晚期腫瘤具有顯著療效而備受關注,它通過激活體內的免疫細胞,特意性地清除癌變的細胞。這種治療方法具有特異性強,作用期長和副作用小等優點,一直以來被認為是治愈腫瘤的終極手段。腫瘤免疫療法在2013年被science評為年度十大科技突破之首之后,腫瘤免疫療法的發展得到更廣泛關注。
腫瘤免疫治療是繼手術治療、藥物治療、放射治療之后的一種新的腫瘤治療手段,在預防腫瘤復發、轉移、治療腫瘤病灶等方面具有其獨特的優勢。世界上首個腫瘤治療性疫苗,即表皮生長因子(egf)腫瘤治療性疫苗,該疫苗可以有效延長晚期肺癌患者的生存期,副反應很輕,且在停藥后自動消失,也未發生自身免疫反應癥狀。可見,腫瘤治療性疫苗在抗腫瘤治療具有明顯的優勢。現有抗腫瘤免疫治療的研究主要集中在提高機體抗腫瘤免疫應答或者阻止腫瘤免疫逃逸,而腫瘤免疫響應是一個相當復雜機制,單純一方面的考慮是難以達到抗腫瘤治療的目的。
針對腫瘤的特異性免疫應答,首先要求機體的抗原呈遞細胞能有效加工和呈遞腫瘤抗原,尤其是腫瘤細胞的免疫原性弱,數量少,更不利于有效的抗原呈遞和效應細胞激活。因此,抗原呈遞細胞的加工水平亟待提高。
有研究表明,抗腫瘤疫苗的免疫效應在體內中不甚理想的主要原因是質粒dna或多肽在進入細胞前就被大量的降解,且腫瘤患者的未成熟樹突狀細胞(dendriticcells,dc)對抗原捕獲的攝取能力較低,這也是為何現有許多抗腫瘤dna或多肽疫苗無法真正進入臨床的主要原因之一。隨著納米技術的不斷發展,已有微球、脂質體、納米粒、囊泡等納米給藥載體包裹dna、多肽疫苗進行免疫研究的報道,納米給藥系統能增加dc對dna或多肽的攝取率,且納米給藥載體本身可以作為佐劑誘導增加t細胞反應而增強免疫效應。
細胞自噬是細胞一種“自食”的過程,依靠溶酶體將細胞內受損、變性或衰老的蛋白質以及細胞器運輸到溶酶體進行消化降解以維持細胞穩態。自噬在機體固有免疫和獲得性免疫中發揮了重要作用,一方面通過溶酶體清除胞質內微生物,發揮固有免疫調控作用;另一方面可以通過調節抗原呈遞,在獲得性免疫反應中發揮效應。
因此,在本領域中期望能夠得到一種將上述兩種作用結合起來,通過調節自噬的過程來調節抗原呈遞的藥物體系。
技術實現要素:
針對現有技術的不足,本發明的目的在于提供一種藥物及其制備方法和應用,所述藥物通過增強自噬來提高抗原呈遞,從而進行腫瘤的免疫治療。
為達到此發明目的,本發明采用以下技術方案:
一方面,本發明提供一種藥物,所述藥物包括細胞自噬誘導分子、腫瘤抗原分子和載體,所述自噬誘導分子和所述腫瘤抗原分子連接在所述載體上。
本發明中,所述細胞自噬誘導分子指的是具有自噬誘導或提升自噬水平的分子,包括具有生物活性的基因,多肽或小分子,所述腫瘤抗原分子指的是腫瘤發生、發展過程中新出現或過度表達的抗原物質,主要指的是蛋白類腫瘤抗原分子。
本發明通過將所述細胞自噬誘導分子和所述腫瘤抗原分子連接在載體上,所述細胞自噬誘導分子通過誘導自噬發生提高樹突狀細胞的自噬水平,加強了樹突狀細胞內的蛋白加工等相關過程,所述腫瘤抗原分子通過載體也被樹突狀細胞高效的攝取到細胞內部,伴隨自噬過程的發生,所述腫瘤抗原分子在樹突狀細胞內部也被有效加工進而呈遞到細胞表面,所述細胞自噬誘導分子和所述腫瘤抗原分子通過連接到同一個載體上,具有協同增效的作用,兩者的效果都比單獨與載體連接后分別進入細胞內部的效果明顯增強。
優選地,所述細胞自噬誘導分子為細胞自噬誘導多肽和/或小分子細胞自噬誘導劑,優選為細胞自噬誘導多肽。
優選地,所述細胞自噬誘導多肽包括beclin1和/或atg蛋白,優選為beclin1。
優選地,所述beclin1的氨基酸序列如seqno.1所示,所述seqno.1的氨基酸序列如下:cys-gly-thr-asn-val-phe-asn-ala-thr-phe-his-ser-gly-gln-phe-gly-thr。
優選地,所述小分子細胞自噬誘導劑包括雷帕霉素和/或鈣蛋白酶抑制劑。
優選地,所述腫瘤抗原分子為模式抗原肽和/或腫瘤表面標志物,優選為模式抗原肽。
優選地,所述模式抗原肽包括卵清蛋白(ova)、附膜蛋白(muc1)或甲胎蛋白(afp)中的任意一種或至少兩種的組合,優選為卵清蛋白。
優選地,所述卵清蛋白的氨基酸序列如seqno.2所示,所述seqno.2的氨基酸序列如下:cys-ser-ile-ile-asn-phe-glu-lys-leu。
優選地,所述腫瘤表面標志物包括附膜蛋白。
本發明中,所述自噬誘導分子和所述腫瘤抗原分子還可以進行靶向性質或親水性質的修飾,本領域技術人員可以根據需要進行這些修飾,在此不做特殊限定,而且靶向性質或親水性質的修飾不會對本申請的治療效果產生影響。
優選地,所述載體為無機金屬載體材料、無機非金屬載體材料或有機高分子材料中的任意一種或至少兩種的組合,優選為有機高分子材料。
優選地,所述無機金屬載體材料包括金納米顆粒、銀納米顆粒或四氧化三鐵納米顆粒中的任意一種或至少兩種的組合。
優選地,所述無機非金屬載體材料包括介孔硅、碳納米管和石墨烯中的任意一種或至少兩種的組合。
優選地,所述有機高分子材料包括聚氨基酯材料,聚乳酸聚乙醇共聚物或脂質體中的任意一種或至少兩種的組合,優選為聚氨基酯材料,進一步優選為修飾了酸敏感側鏈基團的聚氨基酯材料。
本發明中,所述修飾了酸敏感側鏈基團的聚氨基酯材料在溶酶體逃逸階段的酸性調節下,所述聚氨基酯材料結構會變松散,利于藥物的釋放。
優選地,所述聚氨基酯材料的分子式如式i所示:
其中,所述m,n獨立地選自3-40中的正整數,m和n可根據聚合反應時間進行調節,在此不做特殊限定,優選為n=36,m=4。
作為優選技術方案,所述藥物的分子式ii如式所示:
第二方面,本發明提供一種如第一方面所述的藥物的制備方法,包括以下步驟:
(1)制備載體;
(2)將步驟(1)所述載體溶解于有機溶劑中,與所述細胞自噬誘導分子和所述腫瘤抗原分子避光反應;
(3)將步驟(2)反應后的產物用透析袋透析,收集產物即為所述藥物。
優選地,步驟(1)所述的載體為聚氨基酯材料,具體包括:將1,6-己二醇二丙烯酸酯、n,n-二丁基丙二胺和nh2-mpeg2000溶解在有機溶劑中,溶解后通入n2,加熱后反應,將反應物進行透析,得到所述載體,其中,m選自3-40中的正整數,優選為4。
優選地,所述1,6-己二醇二丙烯酸酯、n,n-二丁基丙二胺和nh2-mpeg2000的摩爾比為(8-15):(3-12):1,例如可以是8:3:1、9:3:1、10:3:1、11:3:1、12:3:1、13:3:1、14:3:1、15:3:1、8:4:1、8:5:1、8:6:1、8:7:1、8:8:1、8:9:1、8:10:1、8:11:1、8:12:1、9:4:1、9:5:1、9:6:1、9:7:1、9:8:1、9:9:1、9:10:1、9:11:1、9:12:1、10:4:1、10:5:1、11:5:1、11:6:1、11:7:1、11:8:1、11:9:1、11:10:1、11:12:1、12:4:1、12:6:1、12:8:1、12:10:1、12:12:1、13:4:1、13:6:1、13:8:1、13:10:1、13:12:1、14:4:1、14:6:1、14:8:1、14:10:1、14:12:1、15:4:1、15:6:1、15:8:1、15:10:1、15:12:1,優選為(10-13):(6-10):1,進一步優選為12:9:1。
優選地,所述有機溶劑選自但不限于二甲基亞砜和/或二甲基甲酰胺,優選為二甲基亞砜。
優選地,所述通入n2的時間為10-20min,例如可以是10min、11min、12min、13min、14min、15min、16min、17min、18min、19min或20min,優選為12-16min,進一步優選為15min。
優選地,所述加熱的溫度為40-60℃,例如可以是40℃、41℃、42℃、43℃、44℃、45℃、46℃、47℃、48℃、49℃、50℃、51℃、52℃、53℃、54℃、55℃、56℃、57℃、58℃、59℃或60℃,優選為43-56℃,進一步優選為50℃。
優選地,所述反應的時間為5-10天,例如可以是5天、6天、7天、8天、9天或10天,優選為6-8天,進一步優選為7天。
優選地,所述透析的透析袋的截留分子量為3000-4000,例如可以是3000、3100、3200、3300、3400、3500、3600、3700、3800、3900或4000,優選為3200-3800,進一步優選為3500。
優選地,所述透析的時間為5-10h,例如可以5h、6h、7h、8h、9h或10h,優選為6-9h。
優選地,步驟(2)所述細胞自噬誘導分子和所述腫瘤抗原分子需要過量添加,即所述自噬誘導分子與所述載體的摩爾比不小于1:1,所述腫瘤抗原分子與所述載體的摩爾比不小于1:1。
優選地,步驟(2)所述有機溶劑選自但不限于二甲基亞砜和/或二甲基甲酰胺,優選為二甲基亞砜。
優選地,步驟(2)所述反應的溫度為30-40℃,例如可以是30℃、31℃、32℃、33℃、34℃、35℃、36℃、37℃、38℃、39℃或40℃,優選為35-39℃,進一步優選為37℃。
優選地,步驟(2)所述反應的時間為3-5天,例如可以是3天、4天或5天,優選為3天。
優選地,步驟(3)所述透析袋的截留分子量為3000-4000,例如可以是3000、3100、3200、3300、3400、3500、3600、3700、3800、3900或4000,優選為3200-3800,進一步優選為3500。
優選地,步驟(3)所述透析的時間為5-10h,例如可以5h、6h、7h、8h、9h或10h,優選為6-9h。
作為優選技術方案,所述制備方法包括如下步驟:
(1)將1,6-己二醇二丙烯酸酯、n,n-二丁基丙二胺和nh2-mpeg2000溶解在dmso中,所述1,6-己二醇二丙烯酸酯、n,n-二丁基丙二胺和nh2-mpeg2000的摩爾比為(8-15):(3-12):1,溶解后通入n210-20min,加熱到40-60℃后反應5-10天,將反應物采用截留分子量為3000-4000的透析袋進行透析5-10h,得到所述載體,其中,所述m選自3-40中的正整數,優選為4;
(2)將步驟(1)所述載體溶解于dmso中,加入所述細胞自噬誘導分子和所述腫瘤抗原分子,所述細胞自噬誘導分子與所述載體的摩爾比不小于1:1,所述腫瘤抗原分子與所述載體的摩爾比不小于1:1,30-40℃避光反應3-5天;
(3)將步驟(2)反應后的產物采用截留分子量為3000-4000的透析袋進行透析5-10h,冷凍干燥,即為所述藥物。
第三方面,本發明提供如第一方面所述的藥物在制備腫瘤免疫治療藥物中的應用。
優選地,所述藥物的用量應小于半毒性劑量(<ic50)。
本發明所述分子式ii藥物的用量為60-80μg/ml,例如可以是60μg/ml、61μg/ml、62μg/ml、63μg/ml、64μg/ml、65μg/ml、66μg/ml、67μg/ml、68μg/ml、69μg/ml、70μg/ml、71μg/ml、72μg/ml、73μg/ml、74μg/ml、75μg/ml、76μg/ml、77μg/ml、78μg/ml、79μg/ml或80μg/ml,優選為65μg/ml,選擇在此用量范圍內不會對細胞造成殺傷作用,而是通過刺激細胞調節細胞自身的免疫效果,從而進行腫瘤的治療。
相對于現有技術,本發明具有以下有益效果:
(1)本發明通過將所述細胞自噬誘導分子和所述腫瘤抗原分子連接在同一個載體上,具有協同增效的作用,兩者的效果都比單獨與載體連接后分別進入細胞內部的效果明顯增強,有效誘導自噬從而增強抗原呈遞,最終達到提高治療腫瘤效果的目的;
(2)本發明藥物通過增強自噬從而提高抗原呈遞的藥物為發展腫瘤免疫治療提供了新思路。
附圖說明
圖1是本發明藥物的示意圖;
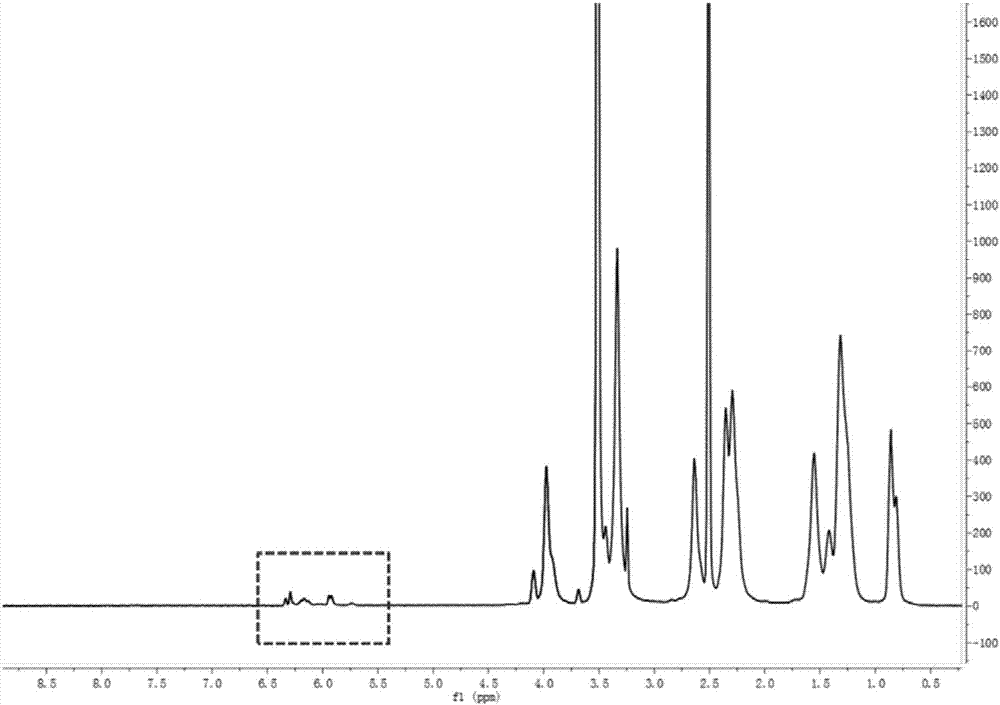
圖2是實施例1中所述載體的核磁圖譜(1hnmr);
圖3是實施例2中所述藥物的核磁圖譜(1hnmr);
圖4是所述藥物分子在加入dc2.4細胞后誘導自噬的共聚焦成像,其中,分子1-實施例1制備的載體,分子2-本發明對比例1制備的對照藥物,分子3-本發明實施例2制備的藥物,分子3+3-ma-分子3與自噬抑制劑3-甲基腺嘌呤,分子2+rapa-分子2和化學自噬誘導劑雷帕霉素;
圖5是實施例1中所述分子加入dc2.4細胞后誘導細胞自噬的蛋白表達變化,其中,圖5(a)為自噬相關蛋白微管相關蛋白1輕鏈3(lc3)相對肌動蛋白actin的表達,圖5(b)為自噬底物相關蛋白泛素結合蛋白p62相對肌動蛋白actin的表達,其中,分子1-實施例1制備的載體,分子2-本發明對比例1制備的對照藥物,分子3-本發明實施例2制備的藥物;
圖6是實施例1中所述分子通過誘導小鼠骨髓來源樹突狀細胞不同程度的自噬后流式檢測細胞表達的特異性抗原水平,其中,分子1-實施例1制備的載體,分子2-本發明對比例1制備的對照藥物,分子3-本發明實施例2制備的藥物,分子3+3-ma-分子3與自噬抑制劑3-甲基腺嘌呤,分子2+rapa-分子2和化學自噬誘導劑雷帕霉素;
圖7是實施例1中所述分子進行腫瘤治療過程中小鼠腫瘤的體積大小,其中,分子1-實施例1制備的載體,分子2-本發明對比例1制備的對照藥物,分子3-本發明實施例2制備的藥物;
圖8是實施例1中所述分子進行腫瘤治療過程中,通過流式細胞術檢測小鼠的腫瘤內部cd8+細胞數量,其中,分子1-實施例1制備的載體,分子2-本發明對比例1制備的對照藥物,分子3-本發明實施例2制備的藥物。
具體實施方式
下面通過具體實施方式來進一步說明本發明的技術方案。本領域技術人員應該明了,所述實施例僅僅是幫助理解本發明,不應視為對本發明的具體限制。
實施例1載體的制備
以聚氨基酯化合物為例,其分子結構為:
聚合物合成具體方法為:(1)稱取1.2mmol的1,6-己二醇二丙烯酸酯,0.9mmol的n,n-二丁基丙二胺以及0.1mmol的nh2-mpeg2000,并全部溶解分散在2ml的二甲基亞砜(dmso)中,完全溶解后通入n215分鐘,然后加熱到50℃避光反應7天;(2)7天后取出反應產物,使用截留量為3500的透析袋在水中透析6-9個小時,收集透析袋中的產物;(3)冷凍干燥,將產物凍干后收集以備后續實驗。
所述載體的核磁結構如圖2所示,虛線框位置表示雙鍵,可見構建成功所述載體。
實施例2藥物的制備
取實施例1中反應產物溶解于dmso,向其中加入過量的細胞自噬誘導肽beclin1,以及腫瘤抗原肽ova,37℃避光反應3天;(2)3天后取出反應產物,使用截留量為3500的透析袋在水中透析6-9個小時,收集透析袋中的產物;(3)冷凍干燥,將產物凍干后收集以備后續實驗。產物具有自噬誘導功能與腫瘤抗原負載的分子。
所得到的修飾多肽的分子,即所述藥物分子3的化學結構如下:
其示意圖如圖1所示,核磁圖譜如圖3所示,從圖中可以看出原來載體分子中聚合物兩端的雙鍵消失(虛線框表示位置),說明雙鍵與多肽中半胱氨酸(cys)上的巰基(-sh)發生了反應,說明多肽有效的共價連接在了載體分子的兩端。
對比例1所述藥物對照分子2的制備
取實施例1中反應產物溶解于dmso,向其中加入過量的不具有自噬誘導功能的對照多肽beclin1scramble:cys-lys-ile-phe-gly-ser-leu-ala-phe-leu,以及腫瘤抗原肽ova,37℃避光反應3天;(2)3天后取出反應產物,使用截留量為3500的透析袋在水中透析6-9個小時,收集透析袋中的產物;(3)冷凍干燥,將產物凍干后收集以備后續實驗。產物不具有自噬誘導功能分子,具有抗原負載的分子,為對照藥物分子2。
實施例3所述藥物提高誘導自噬
首先,使用dc2.4細胞系為對象,進行分子誘導自噬效應的研究。將dc2.4細胞按5×104接種在共聚焦皿中,在37℃以及5%co2的條件下孵育12小時;利用lipo3000對細胞進行轉染,轉染gfp-lc3質粒,待轉染6小時后將含轉染試劑的培養基換成完全培養基,再培養12-16小時;
向轉染后的細胞中分別加入細胞自噬誘導劑,抑制劑以及對應的分子3或對照分子2,孵育12小時后用pbs潤洗細胞并使用共聚焦顯微鏡觀察細胞的gfp熒光,結果如圖4,通過gfp-lc3熒光可初步判斷分子誘導自噬的水平。
當僅用載體分子即分子1處理細胞的時候,gfp-lc3陽性斑點沒有增加,細胞的熒光沒有明顯的增強;當用修飾了腫瘤抗原肽和細胞自噬誘導對照肽的分子2處理細胞時,細胞的gfp-lc3陽性斑點和細胞的熒光沒有明顯的增加;當用修飾了具有細胞自噬誘導功能的多肽和腫瘤抗原肽的分子3處理細胞時,細胞中的gfp-lc3斑點明顯變多,熒光強度也明顯增強,說明自噬水平確實有提高。當輔以細胞自噬誘導劑rapa和分子2共同處理細胞時,細胞中的gfp-lc3熒光增強,說明自噬水平在誘導劑的作用下得到了提升;當用自噬抑制劑3-ma和分子3共同處理細胞時,細胞的gfp-lc3熒光減弱,說明分子3引起的自噬升高被抑制劑3-ma抑制。
再將dc2.4細胞傳代于六孔板,向細胞中分別加入自噬誘導劑,抑制劑以及對應的分子或對照分子等,12小時后收集細胞并用ripa裂解液裂解,收集細胞裂解液并用bsa進行蛋白定量,然后用于蛋白印跡實驗。結果如圖5,通過蛋白印跡實驗結果可以看出,所述實施例2制備的藥物能有效誘導自噬。
實施例4所述藥物提高抗原呈遞效應
選取小鼠骨髓來源的樹突狀細胞為細胞模型用于研究該分子的抗原呈遞效應。按照lutz法分離并擴增小鼠骨髓來源的樹突狀細胞。到第8天,將樹突狀細胞傳代并按5×105重新鋪板于六孔板中;培養12-16小時后,向其中加入自噬誘導劑,抑制劑以及對應的分子或對照分子等;孵育12小時后分別收集細胞;使用流式抗體h-2kb,cd11c(biolegend)孵育細胞并用bdcallibur儀器檢測;此外,還可以收集的上清運用elisa的手段對細胞因子進行檢測。
通過圖6結果可以看到實施例2所述的藥物分子對自噬的調控有效的增強了抗原的呈遞,當用具有細胞自噬誘導功能的分子3處理細胞時,抗原呈遞水平顯著升高;而用不具有細胞自噬誘導功能的分子2處理細胞時,抗原呈遞水平升高不明顯。同樣,當聯合用分子2和自噬激活劑rapa處理細胞時,抗原呈遞水平顯著升高;而用自噬抑制劑3-ma和分子3處理細胞時,由分子3引起的抗原水平升高被下調。該結果說明自噬水平的高低與抗原呈遞的水平是正相關的,通過提高自噬水平以增強抗原呈遞是一種有效的手段。
實施例5所述藥物提高抗原呈遞用于腫瘤治療
將特異性表達卵清蛋白ova抗原的小鼠黑色素瘤細胞b16-f10-ova接種于小鼠c57bl/6右腿外側。約5-7天后,待小鼠荷瘤大小長到200mm3開始進行腫瘤治療。間隔一日靜脈給藥并檢測腫瘤大小,如圖7,所給藥物為上述方法實施例2制備得到的藥物分子,同時給其他組小鼠給相應的對照藥物,進行為期兩周的治療;并通過流式分析腫瘤內部cd8+細胞數量,結果如圖8,可見所述藥物分子能通過提高抗原呈遞增強t細胞的活化,最終達到提高腫瘤治療效果的目的。
申請人聲明,本發明通過上述實施例來說明本發明的載藥聚合物納米膠束及其制備方法和應用,但本發明并不局限于上述實施例,即不意味著本發明必須依賴上述實施例才能實施。所屬技術領域的技術人員應該明了,對本發明的任何改進,對本發明所選用原料的等效替換及輔助成分的添加、具體方式的選擇等,均落在本發明的保護范圍和公開范圍之內。
sequencelisting
<110>國家納米科學中心
<120>一種藥物及其制備方法和應用
<130>2017
<160>2
<170>patentinversion3.3
<210>1
<211>17
<212>prt
<213>人工合成序列
<400>1
cysglythrasnvalpheasnalathrphehisserglyglnphegly
151015
thr
<210>2
<211>9
<212>prt
<213>人工合成序列
<400>2
cysserileileasnpheglulysleu
15
- 還沒有人留言評論。精彩留言會獲得點贊!