一次顆粒為球形的高純高活性碳酸錳的合成的制作方法
本發明涉及一種球形碳酸錳,特別是一次顆粒為球形的高純高活性碳酸錳的合成。
背景技術:
我國錳礦資源豐富,基礎錳源材料如電解金屬錳、電解二氧化錳、硫酸錳、碳酸錳等錳制品產量居世界前列。但精細化、高附加值的錳制品卻較少。碳酸錳是產量較大的基礎錳源材料,主要用于電子工業的鐵氧體軟磁材料、陶瓷、其他錳鹽制備等。近年來,隨著世界各國對新能源材料的高度重視,鋰電池行業特別是動力鋰電行業得到迅速發展,碳酸錳的需求量越來越大,應用領域在不斷拓展,同時,對碳酸錳的品質也提出了更高的要求。比如要求碳酸錳振實密度大且粒徑分布均勻,純度高特別是鋰電敏感雜質鉀鈉鈣鎂鐵等含量低。這種高品質、精細化碳酸錳產品是制備高品質化學二氧化錳、高品質四氧化三錳及錳酸鋰的關鍵錳源材料,對改善鋰電池的放電容量、循環性能、電化學活性有重要促進作用。硫酸錳沉淀法制碳酸錳,要提高碳酸錳的純度主要通過提高原料硫酸錳的純度來實現,而要獲得粒徑分布好、球形度結構好、性能優良的碳酸錳則需要改善碳酸錳的制備工藝。
球形碳酸錳在電子、醫藥、顏料和催化劑等高新技術領域具有廣泛的應用。用球形碳酸錳制備的粒徑分布均勻的球形鋰離子電池正極材料LiMn2O4,具有較高的放電容量、良好的循環能力和高活性。有研究者發現,由許多細小微晶構成的球形MnCO3顆粒,由于有厚實的外殼層,在去核時外殼具有較強的抗酸分解能力,而且用這些MnCO3制備出來的空心微球具有很強的抗機械力和良好的滲透性。片式多層陶瓷電容器的高層數、薄介質、高可靠的發展趨勢也對瓷料中摻雜的MnCO3材料的形貌和大小提出了更高的要求。超細MnCO3的活性越高,越容易溶入鈦酸鋇(BaTiO3)晶格中而形成較薄殼層的殼芯結構。因此,有關不同形貌和粒度的碳酸錳的研究日益受到人們的重視。
通過對碳酸錳制備過程以及產品的分析,研究出了一種可以制備出高活性碳酸錳的合成方法。本發明通過對碳酸錳合成工藝的探索,制備出了一次顆粒為球形的高活性碳酸錳。
技術實現要素:
本發明的目的是提供一種一次顆粒為球形或者類球形的碳酸錳的制備方法,提高碳酸錳材料的活性、振實密度、加工性能,制備出的球形碳酸錳錳含量高、雜質含量低、粒徑較小。
為解決碳酸錳技術問題,本發明所采用的技術方案是:一種生產一次顆粒為球形的高純高活性碳酸錳的工藝方法,包括以下步驟:
a、將可溶性錳鹽除雜精制,配制濃度為0.1M~5M的錳鹽溶液;
b、將碳酸銨配制成濃度為0.1M~5M的碳酸銨溶液;
c、將碳酸氫銨配制成濃度為0.1M~5M的碳酸氫銨溶液;
d、打開反應器中的攪拌器,向反應器中快速加入步驟a中配制的錳鹽溶液和步驟b中配制的碳酸銨溶液,控制碳錳比為n(C):n(Mn)=5:1~1:1,控制反應溫度為5℃~40℃,反應時間為10min~2h;
e、d步驟完成后,取一定量的碳酸氫銨加入反應釜,控制加入量使得n(C):n(Mn)=0:1~4:1,控制反應釜的溫度為10℃~120℃,反應時間為1h~5h;
f、反應完成后,水洗、抽濾,并進行真空干燥,控制干燥溫度為60℃~150℃,干燥時間為6h~48h。得到一、二次顆粒均為球形、平均粒徑為400nm~1μm,錳含量大于45%(重量百分比)的高活性球形碳酸錳。
根據本發明的上述制備方法中,步驟(a)所述的可溶性錳鹽可以是可溶于水的所有錳鹽,來源于錳礦、電解錳、硫酸錳、硝酸錳、氯化錳等。
上述制備方法中攪拌速度為100r/min~1000r/min。
上述制備方法中步驟a中配制的錳鹽溶液和步驟b中配制的碳酸銨溶液的反應方式為并加、正加、反加中的一種。本發明采用的并加反應方式為將錳鹽溶液和(NH4)2CO3溶液同時加入反應器進行反應;本發明采用的正加反應方式為將(NH4)2CO3溶液加入錳鹽溶液中進行反應;本發明采用的反加反應方式為將錳鹽溶液加入(NH4)2CO3溶液中進行反應。
本發明的優點及有益效果:
1)本發明提供的一種生產一次顆粒為球形碳酸錳的工藝方法,生產出的碳酸錳一、二次顆粒均為球形,活性高。
2)本發明制備出的一次顆粒為球形的碳酸錳錳含量高、雜質含量低,錳含量大于45%(重量百分比),高價錳含量低。
3)本發明制備出的一次顆粒為球形的碳酸錳粒徑小平均粒徑為400nm~1μm,粒徑分布窄,D10/D90小于1。
4)本發明工藝方法簡單,pH值影響較小,無需在線監控pH值,適合于大批量生產。
附圖說明
圖1是實施例一制備出的樣品的掃描電鏡圖。
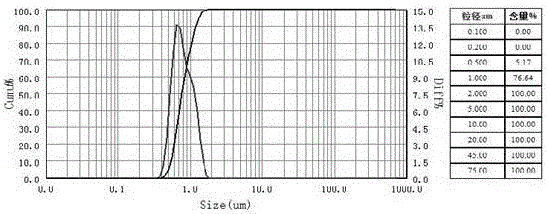
圖2是實施例一制備出的樣品的激光粒度測試分布圖。
圖3是實施例十制備出的樣品的掃描電鏡圖。
圖4是實施例十制備出的樣品的激光粒度測試分布圖。
圖5是實施例十三制備出的樣品的掃描電鏡圖。
圖6是實施例十三制備出的樣品的激光粒度測試分布圖。
具體實施方式
實施例一
稱取96.06g(NH4)2CO3定容至1L,稱取48.03g(NH4)2CO3定容至500mL,配制成1.0M的(NH4)2CO3溶液2.5L;稱取169.016g MnSO4·H2O定容至1L,取84.508g MnSO4·H2O定容至500mL,配制成1.0M的MnSO4·H2O溶液2.5L;打開電機,調節轉速為200r/min,將2.5LMnSO4·H2O溶液和2.5L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為40min,再將反應釜置于85℃水浴中加熱1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于100℃真空干燥箱中干燥10h。
實施例二
稱取96.06g(NH4)2CO3定容至1L,稱取48.03g(NH4)2CO3定容至500mL,配制成1.0M的(NH4)2CO3溶液2.5L;稱取169.016g MnSO4·H2O定容至1L,取84.508g MnSO4·H2O定容至500mL,配制成1.0M的MnSO4·H2O溶液2.5L;打開電機,調節轉速為200r/min,將2.5LMnSO4·H2O溶液和2.5L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為40min,再將反應釜置于85℃水浴中加熱1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于120℃真空干燥箱中干燥10h。
實施例三
稱取96.06g(NH4)2CO3定容至1L,稱取48.03g(NH4)2CO3定容至500mL,配制成1.0M的(NH4)2CO3溶液2.5L;稱取169.016g MnSO4·H2O定容至1L,取84.508g MnSO4·H2O定容至500mL,配制成1.0M的MnSO4·H2O溶液2.5L;打開電機,調節轉速為200r/min,將2.5LMnSO4·H2O溶液和2.5L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為40min,再將反應釜置于85℃水浴中加熱1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于60℃真空干燥箱中干燥10h。
實施例四
稱取96.06g(NH4)2CO3定容至1L,稱取48.03g(NH4)2CO3定容至500mL,配制成1.0M的(NH4)2CO3溶液2.5L;稱取169.016g MnSO4·H2O定容至1L,取84.508g MnSO4·H2O定容至500mL,配制成1.0M的MnSO4·H2O溶液2.5L;打開電機,調節轉速為200r/min,將2.5LMnSO4·H2O溶液和2.5L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為40min,再將反應釜置于95℃水浴中加熱1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于100℃真空干燥箱中干燥10h。
實施例五
稱取96.06g(NH4)2CO3定容至1L,配制成1.0M的(NH4)2CO3溶液3L;稱取169.016g MnSO4·H2O定容至1L,配制成1.0M的MnSO4·H2O溶液2L;打開電機,調節轉速為200r/min,將2LMnSO4·H2O溶液和3L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于120℃真空干燥箱中干燥10h。
實施例六
稱取96.06g(NH4)2CO3定容至1L,配制成1.0M的(NH4)2CO3溶液2L;稱取169.016g MnSO4·H2O定容至1L,配制成1.0M的MnSO4·H2O溶液2L;打開電機,調節轉速為200r/min,將2LMnSO4·H2O溶液和3L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于120℃真空干燥箱中干燥10h。
實施例七
稱取96.06g(NH4)2CO3定容至1L,配制成1.0M的(NH4)2CO3溶液3L;稱取169.016g MnSO4·H2O定容至1L,配制成1.0M的MnSO4·H2O溶液2L;打開電機,調節轉速為200r/min,將2.5LMnSO4·H2O溶液和2.5L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為40min,再將反應釜置于95℃水浴中加熱1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于100℃真空干燥箱中干燥10h。
實施例八
稱取96.06g(NH4)2CO3定容至1L,配制成1.0M的(NH4)2CO3 溶液2L;稱取169.016g MnSO4·H2O定容至1L,配制成1.0M的MnSO4·H2O溶液2L;稱取79.06g NH4HCO3定容至1L,配制成1.0M的NH4HCO3溶液2L。打開電機,調節轉速為200r/min,將2L MnSO4·H2O溶液和2L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為40min。完成后,再將2LNH4HCO3溶液加入反應釜中,并開始加熱反應釜至95℃,反應時間為1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于100℃真空干燥箱中干燥10h。
實施例九
稱取96.06g(NH4)2CO3定容至1L,配制成1.0M的(NH4)2CO3 溶液2L;稱取169.016g MnSO4·H2O定容至1L,配制成1.0M的MnSO4·H2O溶液2L;稱取79.06g NH4HCO3定容至1L,配制成1.0M的NH4HCO3溶液4L。打開電機,調節轉速為200r/min,將2L MnSO4·H2O溶液和2L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為40min。完成后,再將4LNH4HCO3溶液加入反應釜中,并開始加熱反應釜至95℃,反應時間為1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于100℃真空干燥箱中干燥10h。
實施例十
稱取96.06g(NH4)2CO3定容至1L,配制成1.0M的(NH4)2CO3 溶液2L;稱取169.016g MnSO4·H2O定容至1L,配制成1.0M的MnSO4·H2O溶液2L;稱取79.06g NH4HCO3定容至1L,配制成1.0M的NH4HCO3溶液4L。打開電機,調節轉速為200r/min,將2L MnSO4·H2O溶液和2L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為40min。完成后,再將4LNH4HCO3溶液加入反應釜中,并開始加熱反應釜至95℃,反應時間為1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于60℃真空干燥箱中干燥10h。
實施例十一
稱取28.82g(NH4)2CO3定容至1L,配制成0.3M的(NH4)2CO3溶液1L;稱取202.82g MnSO4·H2O定容至1L,配制成1.2M的MnSO4·H2O溶液4L;打開電機,調節轉速為200r/min,將2.5LMnSO4·H2O溶液和2.5L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為40min,再將反應釜置于85℃水浴中加熱1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于100℃真空干燥箱中干燥10h。
實施例十二
稱取28.82g(NH4)2CO3定容至1L,配制成0.3M的(NH4)2CO3溶液1L;稱取202.82g MnSO4·H2O定容至1L,配制成1.2M的MnSO4·H2O溶液4L;打開電機,調節轉速為200r/min,將1LMnSO4·H2O溶液和4L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于120℃真空干燥箱中干燥10h。
實施例十三
稱取28.82g(NH4)2CO3定容至1L,配制成0.3M的(NH4)2CO3溶液1L;稱取202.82g MnSO4·H2O定容至1L,配制成1.2M的MnSO4·H2O溶液4L;稱取55.34g NH4HCO3定容至1L,配制成0.7M的NH4HCO3溶液3.42L。打開電機,調節轉速為200r/min,將1L MnSO4·H2O溶液和4L(NH4)2CO3溶液快速加入反應釜中進行沉淀反應,反應時間為40min。完成后,再將3.42L NH4HCO3溶液加入反應釜中,并開始加熱反應釜至95℃,反應時間為1h。反應完成后,將產物過濾,將得到的濾餅取出,置于去離子水中清洗,再過濾,重復洗滌-過濾過程兩遍。將洗滌完的產物置于100℃真空干燥箱中干燥10h。
- 還沒有人留言評論。精彩留言會獲得點贊!