一種可溶性苯并噻吩衍生物及其制備和應用的制作方法
本發明涉及有機半導體材料技術領域,特別涉及一種可溶性苯并噻吩衍生物及其制備和應用。
背景技術:
近年來,隨著有機薄膜晶體管(Organic thin-film transistor,OTFT)在集成電路和傳感器等方面應用的發展,對高遷移率有機半導體材料的研究和開發非常活躍。可溶液法加工是OTFT的獨特優勢之一,采用溶液法制備器件可以大幅降低器件成本,從而有利于器件的廣泛應用,這也使得通過溶液法制備成為OTFT的研究熱點。
并苯噻吩類化合物是一類非常有應用前景的的小分子材料,1998年,劍橋大學Li等首先采用并三噻吩的二聚體作為場效應晶體管的活性層進行了研究,發現其遷移率為0.05cm2V-1s-1左右,但其開關電流比非常高,達108[Li X C,Sirringhaus H,Garnier F,et al.J.Am.Chem.Soc.,1998,120:2206-2207]。2005年,劉云祈等采用并五噻吩作為晶體管的活性層,并對器件進行適度的優化,其場效應性能得到了大幅度的提升,遷移率達到0.045cm2V-1s-1[Xiao K,Liu Y Q,Qi T,et al.J.Am.Chem.Soc.,2005,127:13281-13286]。但是,并噻吩類衍生物的溶解性較差,無法滿足溶液法加工OTFT的要求,大多仍是采用蒸鍍法進行器件的制備。
目前,現有技術已公開了多種可溶性苯并噻吩類衍生物。其中Takimiya等制備了高溶解度的苯并噻吩類衍生物Cn-BTBTs(n=5~14)(參見如下),基于C13-BTBT的OTFTs的空穴遷移率可達17.2cm2V-1s-1[T Izawa,E Miyazaki,K Takimiya.Adv.Mater.,2008,20(18):3388~3392],結合打印制備單晶膜技術,C8-BTBT單晶膜的OTFTs的遷移率最高可達31.3cm2V-1s-1[H Minemawari,T Yamada,H Matsui et al.Nature,2011,475(7356):364~367]。Nakayama等采用旋涂法制備了基于C10-DNTT的薄膜器件,遷移率平均值達到7.0cm2V-1s-1,在塑料柔性襯底上制備的器件遷移率更高達9.0cm2V-1s-1[K Nakayama,Y Hirose,J Soeda et al.Adv.Mater.,2011,23(14):1626~1629]。但是這些已公開的噻吩類衍生物合成工藝苛刻,制備成本較高,目前仍停留在實驗室制備階段,難以滿足工業生產大量使用的需求。
技術實現要素:
本發明的目的在于克服現有技術的不足,提供一種可溶性苯并噻吩衍生物及其制備和應用。
為實現上述目的,本發明采用的技術方案為:
一種可溶性并噻吩衍生物,衍生物如式(1),(2)或(3)結構所示,
式(1),(2)或(3)中,R為氫原子、C1~18烷基、C1~18烷氧基、C1~18鹵代烷氧基、C1~18烷硫基、C1~18鹵代烷硫基、C1~18烷基磺酰基、C1~18鹵代烷基磺酰基、未取代的或被獨立選自以下基團取代的苯基、聯苯基、吡啶基、噻吩基:C1-C10烷基、C1-C10烷氧基或C1-C10鹵代烷氧基。
式(1),(2)或(3)結構所示可溶性并噻吩衍生物是以平面性和共軛性較好的苯并噻吩體系為核心,并在其最外側芳香環上引入多種取代基團,在提高溶解性的同時可盡可能地不破壞其共軛性,實現較高的場效應遷移率。
優選是,所述通式(1),(2)或(3)中R為氫原子、C4-12烷基、C4-12烷氧基、C4-12鹵代烷氧基、C4-12烷硫基、C4-12鹵代烷硫基、C4-12烷基磺酰基、C4-12鹵代烷基磺酰基、未取代的或被獨立選自以下基團取代的苯基、聯苯基、吡啶基、噻吩基:C1-C10烷基、C1-C10烷氧基或C1-C10鹵代烷氧基。
進一步的優選是,所述通式(1),(2)或(3)中R為氫原子、C6-10烷基、C6-10烷氧基、C6-10鹵代烷氧基、C6-10烷硫基、C6-10鹵代烷硫基、C6-10烷基磺酰基、C6-10鹵代烷基磺酰基、未取代的或被獨立選自以下基團取代的苯基、聯苯基、吡啶基、噻吩基:C1-C10烷基、烷氧基或鹵代烷氧基。
更進一步的優選是,式(1),(2)或(3)所示具體化合物為,
一種可溶性苯并噻吩衍生物的制備方法:
式(1)結構所示的可溶性苯并噻吩衍生物的制備方法,將二苯并噻吩硼酸酯化產物與3-R基-6-三異丙基硅基乙炔基溴苯進行Suzuki偶聯反應;然后在四丁基氟化銨(TBAF)的作用下脫掉保護基三異丙基硅;最后在PtCl2的作用下閉環得到如式(1) 結構所示的可溶性苯并噻吩衍生物。
所述硼酸酯化反應時,將二鹵二苯并噻吩與異丙醇頻哪醇硼酸酯以1:2~1:6的摩爾比在溶劑的存在下于-78℃~-50℃進行硼酸酯化反應6~24小時;
反應式如下:(X為鹵原子)
所述Suzuki偶聯反應是,將二苯并噻吩硼酸酯化產物與3-R基-6-三異丙基硅基乙炔基溴苯以1:2~1:6的摩爾比在有機鈀催化劑、堿和有機溶劑的存在下,于20℃~150℃進行Suzuki偶聯反應1~40小時;
所述有機鈀催化劑添加量為硼酸酯化產物物質的量的1%~10%,有機鈀催化劑為四三苯基膦鈀、醋酸鈀、三(二亞芐基丙酮)二鈀、雙三苯基膦二氯化鈀,[1,1'-雙(二苯基膦基)二茂鐵]二氯化鈀中一種或幾種;
堿加入量為硼酸酯化產物物質的量的1~10倍,堿為碳酸鉀,碳酸鈉,碳酸銫,磷酸鉀,醋酸鉀,乙醇鈉,叔丁醇鉀,三乙胺或二異丙基乙基胺;
有機溶劑為甲苯、1,4-二氧六環,四氫呋喃、二氯甲烷,二甲基亞砜,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺,N-甲基吡咯烷酮中一種或幾種。
反應式如下:
所述在TBAF的作用下脫掉保護基三異丙基硅的反應是,將Suzuki偶聯反應產物與TBAF以1:1~1:3的摩爾比在有機溶劑的作用下于-20℃~80℃反應1~40小時。
所述有機溶劑為甲苯、1,4-二氧六環,四氫呋喃、二氯甲烷,二甲基亞砜,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺,N-甲基吡咯烷酮中一種或幾種。
反應式如下:
所述閉環反應是,將在TBAF的作用下脫掉保護基三異丙基硅的反應產物與PtCl2在溶劑的作用下,于-20℃~150℃反應1~40小時。
所述PtCl2添加量為脫掉三異丙基硅的產物物質的量的1%~20%;
所述有機溶劑為甲苯、1,4-二氧六環,四氫呋喃、二氯甲烷,二甲基亞砜,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺,N-甲基吡咯烷酮中一種或幾種;
反應式如下:
式(2)結構所示的可溶性苯并噻吩衍生物的制備方法,將并三噻吩硼酸酯化產物與3-R基-6-三異丙基硅基乙炔基溴苯進行Suzuki偶聯反應;然后在TBAF的作用下脫掉保護基三異丙基硅;最后在PtCl2的作用下閉環得到如式(2)結構所示的苯并噻吩衍生物。
所述硼酸酯化反應時,將二鹵并三噻吩與異丙醇頻哪醇硼酸酯以1:2~1:6的摩爾比在溶劑的存在下于-78℃~-50℃進行硼酸酯化反應6~24小時;
反應式如下:(X為鹵原子)
所述Suzuki偶聯反應是,將并三噻吩硼酸酯化產物與3-R基-6-三異丙基硅基乙炔基溴苯以1:2~1:6的摩爾比在有機鈀催化劑、堿和有機溶劑的存在下,于20℃~150℃進行Suzuki偶聯反應1~40小時。
所述有機鈀催化劑添加量為并三噻吩硼酸酯化產物物質的量的1%~10%,有機鈀催化劑為四三苯基膦鈀、醋酸鈀、三(二亞芐基丙酮)二鈀、雙三苯基膦二氯化鈀,[1,1'-雙(二苯基膦基)二茂鐵]二氯化鈀中一種或幾種;
堿加入量為并三噻吩硼酸酯化產物物質的量的1~10倍,堿為碳酸鉀,碳酸鈉,碳酸銫,磷酸鉀,醋酸鉀,乙醇鈉,叔丁醇鉀,三乙胺或二異丙基乙基胺;
有機溶劑為甲苯、1,4-二氧六環,四氫呋喃、二氯甲烷,二甲基亞砜,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺,N-甲基吡咯烷酮中一種或幾種。
反應式如下:
所述在TBAF的作用下脫掉保護基三異丙基硅的反應是,將Suzuki偶聯反應產物與TBAF以1:1~1:3的摩爾比在有機溶劑的作用下于-20℃~80℃反應1~40小時;
有機溶劑為甲苯、1,4-二氧六環,四氫呋喃、二氯甲烷,二甲基亞砜,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺,N-甲基吡咯烷酮中一種或幾種。
反應式如下:
所述閉環反應是,將在TBAF的作用下脫掉保護基三異丙基硅的反應產物與PtCl2在溶劑的作用下,于-20℃~150℃反應1~40小時;
所述PtCl2添加量為脫掉三異丙基硅的產物物質的量的1%~20%;溶劑為甲苯、1,4-二氧六環,四氫呋喃、二氯甲烷,二甲基亞砜,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺,N-甲基吡咯烷酮中一種或幾種。
反應式如下:
式(3)結構所示的可溶性苯并噻吩衍生物的制備方法,將2,6-二溴并三噻吩經鋰化反應,再經氰基硼氫化鈉還原,還原產物再經叔丁基鋰和N,N-二甲基甲酰胺作用得到醛基物,所得醛基物經Amberlyst反應關環得到如式(3)結構所示的可溶性并苯噻吩衍生物;
所述鋰化反應是2,6-二溴并三噻吩與2-R基-4-甲酰基噻吩在正丁基鋰的作用下在有機溶劑的存在下于-100℃~0℃與正丁基鋰以摩爾比為1:1~1:3的比例進行6~24小時的反應。
有機溶劑為甲苯、1,4-二氧六環,四氫呋喃、二氯甲烷,二甲基亞砜,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺,N-甲基吡咯烷酮中一種或幾種。
反應式如下:
所述氰基硼氫化鈉還原,是鋰化反應產物與氰基硼氫化鈉在有機溶劑作用下,以1:1~1:3的摩爾比進行還原反應1-24小時;
有機溶劑為甲苯、1,4-二氧六環,四氫呋喃、二氯甲烷,二甲基亞砜,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺,N-甲基吡咯烷酮中一種或幾種。
反應式如下:
所述醛基物,是經氰基硼氫化鈉還原的還原產物在有機溶劑存在下于-100℃~0℃與叔丁基鋰以1:1~1:3的摩爾比進行鋰化反應1~10小時后,再與N,N-二甲基甲酰胺于-100℃~60℃反應1~24小時得到的產物。
有機溶劑為甲苯、1,4-二氧六環,四氫呋喃、二氯甲烷,二甲基亞砜,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺,N-甲基吡咯烷酮中一種或幾種。
反應式如下:
所述Amberlyst反應,是所得醛基物在有機溶劑存在下,在Amberlyst15和BF3-Et2O的作用下于-20℃~110℃反應10分鐘~10小時;
有機溶劑為甲苯、1,4-二氧六環,四氫呋喃、二氯甲烷,二甲基亞砜,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺,N-甲基吡咯烷酮中一種或幾種;Amberlyst15加入量為所得醛基物物質的量的10%~50%,BF3-Et2O加入量為所得醛基物物質的量的10%~50%。
反應式如下:
一種有機薄膜晶體管用材料,所述有機薄膜晶體管用材料為通式(1),(2)或(3)所示化合物。
所述通式(1),(2)或(3)所示化合物作為旋涂用材料。
一種有機薄膜晶體管,有機薄膜晶體管為基板、設置有柵極的介電層和兩端分別設置有漏電極和源電極的半導體層,所述半導體層為通式(1),(2)或(3)所示的可溶性并苯化合物;
其中,所述半導體層復合于所述介電層上,所述介電層復合于所述基板上:或者所述介電層復合于所述半導體層上,所述半導體層復合于所述基板上。
所述半導體層是將通式(1),(2)或(3)所示的可溶性并苯化合物經有機溶劑溶解,而后退火、沉積電極,即得到半導體層。
所述有機溶劑為三氯甲烷、三氯乙烷、氯苯、二氯苯、三氯苯、氯代甲苯、甲苯、二甲苯、苯甲醚、四氫萘或三甲苯。
具體是,上述通式(1),(2)或(3)所示的可溶性并苯化合物用于晶體管之類的電子器件中時,使用高純度的上述化合物材料可以得到電場效應遷移率或開/關比高的器件。因此根據需要,優選通過柱色譜、重結晶、蒸餾、升華等方法對上述通式(1),(2)或(3)所示的可溶性并苯化合物進行精制。優選可以將這些精制方法反復使用,或將過個方法組合,由此來提高純度。進而。作為精制的最終工序,優選將升華精制至少重復2次以上。優選通過上述方法得到的HPLC測定的純度為90%以上的材料,進一步優選使用95%以上的材料,特別優選使用99%以上的材料,由此可以提高有機薄膜晶體管的電場效應遷移率或開關電流比,可以發揮材料本來所特有的性能。
本發明的有機半導體材料可作為旋涂用材料來使用。
本發明有機薄膜晶體管,包括基板、設置有柵極的介電層和兩端分別設置有漏電極和源電極的半導體層,所述半導體層包含上述所述的可溶性并噻吩衍生物;其中,所述半導體層復合于所述介電層上,所述介電層復合于所述基板上:或者所述介電層復合于所述半導體層上,所述半導體層復合于所述基板上。
本發明中,所述有機薄膜晶體管(OTFT)包括基板。所述基本為本領域常用的基本,可以是硅片、玻璃或塑料薄片。對于制備柔性器件,所述有機薄膜晶體管常采用塑料基板,如聚酯(polyester)、聚碳酸酯(polycarbonate)或聚酰亞胺(polyimide)等材料。本發明對所述基板的厚度沒有特殊限制,一般為10微米~10毫米,對于柔性塑料基板,其厚度優選為50微米~5毫米,對于硅片的硬質基板,其厚度優選為0.5毫米~10毫米。
所述有機薄膜晶體管包括介電層,其設置有柵極。所述柵極由導電材料形成,可以是金屬薄膜、導電聚合物薄膜、由導電墨水或導電膠形成的導電薄膜或基板自身如重摻雜的硅片。其中,所述金屬薄膜可以是鋁、金、銀、鉻或氧化銦錫(ITO);所述導電聚合物薄膜可以是聚(苯乙烯磺酸鹽)摻雜的聚(3,4-二氧乙烷噻吩)(PEDOT:PSS);所述導電墨水可以是碳黑(carbon black);所述導電膠可以是銀膠(silver colloid)。所述柵極的厚度可根據所用材料而定,對于由金屬薄膜形成的柵極,其厚度一般為10納米~100納米;對于由導電聚合物形成的柵極,其厚度一般為0.5微米~10微米。
所述介電層通常由無機材料、有機聚合物或有機聚合物和無機材料雜化材料的薄膜形成。其中,所述無機材料為二氧化硅、氮化硅、氧化鋁、鈦酸鋇、鎬酸鋇或五氧化二鉭;所述有機聚合物為聚甲基丙烯酸甲酯(PMMA)、聚乙烯基苯酚(PVP)、聚乙烯醇(PVA)、聚苯乙烯(PS)、聚氯乙烯(PVC)或聚酰亞胺。所述介電層的厚度依賴于所用材料的介電常數,一般為10納米~500納米。
本發明可以采用修飾劑選擇性地所述介電層進行修飾,以形成修飾層,來改變介電層與半導體層間的界面性質,有益于提高有機薄膜晶體管器件的性能。所述修飾劑包括含硅化合物、含磷酸化合物和高介電常數聚合物等。其中,所述含硅化合物可與介電層上得自由羥基發生化學反應,廣泛應用于介電層的自組裝單層(slef-assemble monolayer)修飾;常用的含硅化合物包括十八烷基三氯硅烷(ODTS)、苯基三氯硅烷和含氟烷基三氯硅烷等,具體的含硅化合物修飾劑和修飾方法可參考應用物理雜質(J.Appl.Phys.,2004,96,6431-6438)的相關描述。所述含磷酸化合物也可應用于介電層的自組裝單層修飾;常用的含磷酸化合物包括碳鏈長度12~16的磷酸和苯基取代的磷酸等,具體的含磷酸化合物修飾劑和修飾方法可參考物理化學雜志B輯(J.Phys.Chem.B,2003,107,5877-5881)的相關描述。所述高介電常數聚合物包括聚甲基丙烯酸甲酯(PMMA)、聚乙烯基苯酚(PVP)、聚乙烯醇(PVA)、聚苯乙烯(PS)、聚氯乙烯(PVC)或聚酰亞胺等,具體的種類可參考先進材料雜志(Adv.Mater.,2005,17,1705-1725)的相關描述。
所述有機薄膜晶體管包括半導體層,其兩端分別設置有漏電極和源電極。所述源電極和所述漏電極均可以采用與所述柵極相同的材料,但要保證電極材料與半導體層材料間有小的接觸電阻。本發明對所述漏電極和所述源電極的厚度等沒有特殊限制,其厚度優選為40納米~100納米,形成的導電溝道的寬長比優選為30。
所述半導體層包含上文所述的具有式(1),(2),(3)結構的可溶性并苯噻吩衍生物,所述可溶性并苯噻吩衍生物在有機溶劑中具有較好的溶解性,采用溶液法很容易加工成膜,從而能簡化有機薄膜晶體管的制備方法。例如,相對于并三噻吩,本發明具有式(1),(2),(3)結構的可溶性并苯噻吩衍生物的溶解性至少有100%的提高。在本發明中,所述有機溶劑可以為三氯甲烷、三氯乙烷、氯苯、二氯苯、三氯苯、氯代甲苯、甲苯、二甲苯、苯甲醚、四氫萘或三甲苯。其中,三氯甲烷、三氯乙烷、氯苯、二氯苯、三氯苯或氯代甲苯等屬于氯代溶劑,所述可溶性并苯噻吩衍生物在甲苯溶劑中的溶解度一般為0.05wt%~5wt%。
同時,所述可溶性并苯噻吩衍生物能提高有機薄膜晶體管的載流子遷移率,利于應用。所述半導體層優選還包含聚合物,即所述可溶性并苯噻吩衍生物與聚合物共混形成 半導體層。所述聚合物包括含三芳胺聚合物(poly(triarylamine))、聚咔唑、聚芴、聚噻吩、聚乙烯、聚苯乙烯、聚甲基丙烯酸甲酯、聚乙烯基苯酚和聚碳酸酯等。
在本發明中,半導體層的制備方法優選具體為:將所述可溶性并苯噻吩衍生物配成溶液后制備薄膜,經退火、沉積電極,得到半導體層。
在本發明中,所述可溶性并苯噻吩衍生物配成溶液后制備薄膜的加工技術包括旋涂(spin-coating)、浸涂(dip-coating)、刮涂(blade-coating)、絲網印刷(screening-printing)、噴墨打印(inkjet-printing)等現階段常用的溶液法成膜技術。所述薄膜的厚度優選控制為10納米~100納米,更優選控制為30納米~60納米。所述退火的溫度優選為50℃~150℃,更優選為80℃~120℃;所述退火的時間優選為10分鐘~50分鐘,更優選為20分鐘~40分鐘。
在本發明中,所述半導體層復合于所述介電層上,所述介電層復合于所述基本上;或者所述介電層復合于所述半導體層上,所述半導體層復合于所述基板上。另外,所述介電層選擇性地包括修飾層。
上述烷基,可舉出甲基、乙基、丙基、異丙基、正丁基、仲丁基、異丁基、叔丁基、正戊基、正庚基、正辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基、正十八烷基等。
上述鹵代烷基,例如可舉出氯甲基、1-氯乙基、2-氯乙基、2-氯異丁基、1,2-二氯乙基、1,3-二氯異丙基、2,3-二氯-叔丁基、1,2,3-三氯丙基、1,2,3-三溴丙基、碘甲基、1-碘乙基、2-碘乙基、2-碘異丁基、1,2-二碘乙基、1,3-二碘異丙基、2,3-二碘-叔丁基、1,2,3-三碘丙基、氟甲苯、1-氟甲基、2-氟甲基、2-氟異丁苯、1,2-二氟乙基、二氟甲基、三氟甲基、五氟乙基、全氟異丙基、全氟丁基、全氟環己基等。
上述烷氧基為OY1所表示的基團,Y1可舉出與上述烷基中說明的基團相同的例子,上述鹵代烷氧基是-OY2所表示的基團,Y2可舉出與上述鹵代烷基中說明的基團相同的例子。
上述烷硫基是-SY1所表示的基團,Y1可舉出與上述烷基中說明的基團相同的例子,上述鹵代烷硫基是-SY2所表示的基團,Y2可舉出與上述鹵代烷基中說明的基團相同的例子。
上述烷基磺酰基是-SO2Y1所表示的基團,Y1可舉出與上述烷基中說明的基團同樣的例子,上述鹵代烷基磺酰基為所-SO2Y2所表示的基團,Y2可舉出與上述鹵代烷基中說明的基團相同的例子。
以下,舉出本發明的有機薄膜晶體管用化合物的具體例,本發明不限定于下述化合物。
本發明所具有的優點:
與現有技術相比,本發明提供的可溶性并苯噻吩衍生物具有式(1),(2)或(3)的結構。本發明向雜環體系中引入不同碳鏈長度的烷基、烷氧基、烷硫基等不同類型的取代基,采用這種對并三噻吩的選擇性取代,能夠在提高溶解性的同時盡可能地不破壞并苯噻吩的共軛性,可以降低取代基對并并苯噻吩核在薄膜中的排列方式的不良影響,進而實現較高的場效應遷移率。因此,采用本發明提供的可溶性并苯噻吩衍生物能夠獲得具有較高遷移率的有機薄膜晶體管。實驗結果表明,采用本發明提供的可溶性并噻吩衍生物在常規有機溶劑的溶解度達到50mg/ml,滿足溶液法制備薄膜晶體管的要求,所制備的有機薄膜晶體管,其載流子遷移率可達到0.62cm2V-1s-1,開關比達106。
另外,本發明豐富了可溶性并噻吩衍生物的種類,而且本發明采用可溶性并噻吩衍生物以溶液沉積的方法制備有機薄膜晶體管,方法簡便、成本較低。
附圖說明
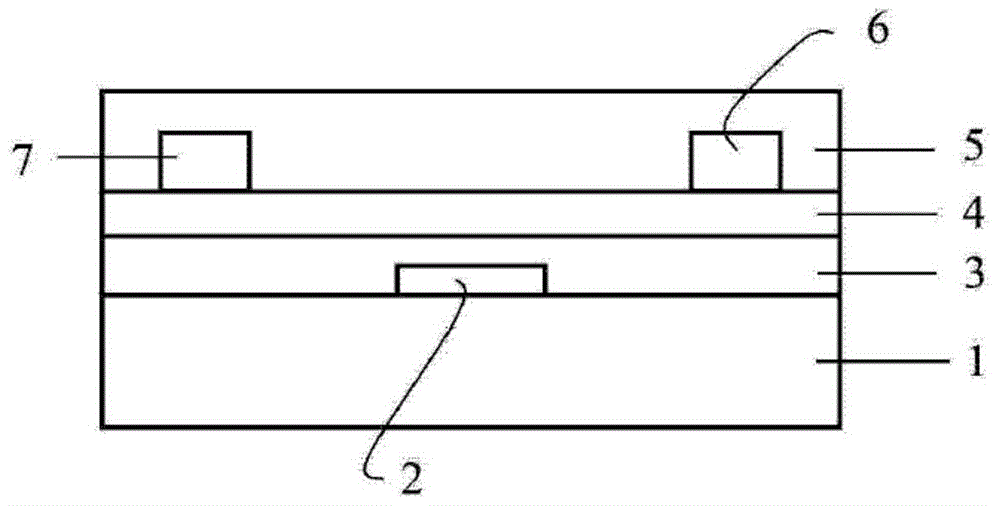
圖1為本發明實施例提供的的有機薄膜晶體管的第一種結構示意圖;其中,1為基 板,2為柵極,3為介電層,4為修飾層,5為半導體層,6為源電極,7為漏電極;半導體層5復合于修飾層4上,修飾層4復合于介電層3上,介電層3復合于基板1上;柵極2設置于介電層3上,源電極6和漏電極7設置于半導體層5兩端的上表面。
圖2為本發明實施例提供的有機薄膜晶體管的第二種結構示意圖;其中,圖2與圖1的區別僅在于,源電極6和漏電極7分別設置于半導體層5兩端的下表面。
圖3為本發明實施例提供的有機薄膜晶體管的第三種結構示意圖,其中,介電層3復合于半導體層5上,半導體層5復合于修飾層4上,修飾層4復合于基板1上;柵極2設置于介電層3上,源電極6和漏電極7分別設置于半導體層5兩端的下表面。
具體實施方式
為了進一步理解本發明,下面結合實施例對本發明優選實施方案進行描述,但是應當理解,這些描述只是為進一步說明本發明的特征和有點,而不是對發明權利要求的限制。
實施例1化合物(A-3)的制備
在干燥的并持續通入氮氣的反應器中加入3,7-二硼酸酯基二苯并噻吩(2.18g,5mmol)、3-正己基-6-三異丙基硅基乙炔基溴苯(4.5g,11mmol)、四三苯基膦鈀(0.58g,0.5mmol)、無水碳酸鉀(2.07g,15mmol)、甲苯(50ml)、水(5ml),緩慢升溫至回流,攪拌24小時。然后將反應溶液倒入100ml稀鹽酸(10%)中。用乙醚萃取兩次,合并有機相用水洗一次,再用飽和食鹽水洗一次,用無水硫酸鈉干燥有機相,過濾后減壓蒸出有機溶劑,并用石油醚柱層析得到淡黃色油狀液體化合物1(2.3g,收率53%)。
向燒瓶中加入化合物1(2.16g,2.5mmol)、四丁基氟化銨(1.24g,5mmol)甲醇(20ml)、二氯甲烷(20ml),室溫攪拌16h,加水和乙醚萃取,然后再用乙醚萃取兩次,合并有機相用水洗一次,再用飽和食鹽水洗一次,用無水硫酸鈉干燥有機相,過濾后減壓蒸出有機溶劑,并用石油醚柱層析得到淡黃色油狀液體(0.58g,收率42%)
在干燥的并持續通入氮氣的反應器中,在避光條件下加入化合物2(1.1g,2mmol)、干燥的四氫呋喃(20ml)。氮氣鼓泡15分鐘,然后向溶液中加入PtCl2(266mg,5%mol)繼續鼓泡15分鐘。反應溶液在室溫下攪拌,反應16小時。用2cm高的硅膠過濾反應液,減壓蒸餾除去濾液溶劑,粗產品利用柱層析進行提純(石油醚),再用甲苯重結晶,得到淡黃色晶體化合物A-3(0.57g,收率52%)。
實施例2化合物(A-7)的制備
與實施例1不同之處在于,將3-正己基-6-三異丙基硅基乙炔基溴苯用3-正癸基-6-三異丙基硅基乙炔基溴苯替換,而后按照實施例1的步驟制備獲得化合物(A-7),如下式所示。
實施例3化合物(B-3)的制備
在干燥的并持續通入氮氣的反應器中加入2,6-二硼酸酯基三噻吩(2.24g,5mmol)、3-正己基-6-三異丙基硅基乙炔基溴苯(4.6g,11mmol)、四三苯基膦鈀(0.58g,0.5mmol)、無水碳酸鉀(2.07g,15mmol)、甲苯(50ml)、水(5ml),緩慢升溫至回流,攪拌24小時。然后將反應溶液倒入100ml稀鹽酸(10%)中。用乙醚萃取兩次,合并有機相用水洗一次,再用飽和食鹽水洗一次,用無水硫酸鈉干燥有機相,過濾后減壓蒸出有機溶劑,并用石油醚柱層析得到淡黃色油狀液體化合物4(2.7g,收率61.5%)。
向燒瓶中加入化合物4(2.19g,2.5mmol)、四丁基氟化銨(1.24g,5mmol)甲醇(20ml)、二氯甲烷(20ml),室溫攪拌16h,加水和乙醚萃取,然后再用乙醚萃取兩次,合并有機相用水洗一次,再用飽和食鹽水洗一次,用無水硫酸鈉干燥有機相,過濾后減壓蒸出有機溶劑,并用石油醚柱層析得到淡黃色油狀液體化合物5(0.52g,收率37%)
在干燥的并持續通入氮氣的反應器中,在避光條件下加入化合物5(1.13g,2mmol)、干燥的四氫呋喃(20ml)。氮氣鼓泡15分鐘,然后向溶液中加入PtCl2(266mg,5%mol)繼續鼓泡15分鐘。反應溶液在室溫下攪拌,反應16小時。用2cm高的硅膠過濾反應液,減壓蒸餾除去濾液溶劑,粗產品利用柱層析進行提純(石油醚),再用甲苯重結晶,得到淡黃色晶體化合物B-3(0.54g,收率48%)。
實施例4化合物(B-5)的制備
與實施例3不同之處在于,將3-正己基-6-三異丙基硅基乙炔基溴苯用3-正辛基-6-三異丙基硅基乙炔基溴苯替換,而后按照實施例3的步驟制備獲得化合物(B-5),如下式所示。
實施例5化合物(C-1)的制備
在干燥的并持續通入氮氣的反應器中下加入化合物8(2.83g,8mmol)、干燥的四氫呋喃(40ml)。降溫至-78℃,然后向溶液中緩慢滴加1.6M的n-BuLi(12ml,9.6mmol)。反應溶液在-78℃下攪拌,反應1.5小時。開始滴加化合物9的無水THF溶液(3.75g/20ml,15.2mmol)。滴加完畢后使反應液升溫至室溫繼續反應20h,用水萃滅反應后,用乙醚萃取兩次,合并有機相用水洗一次,再用飽和食鹽水洗一次,用無水硫酸鈉干燥有機相,過濾后減壓蒸出有機溶劑,并用石油醚柱層析得到淡黃色油狀液體化合物10(2.2g,收率42%)
向燒瓶中加入化合物10(1.38g,2mmol)、氰基硼氫化鈉(189mg,3mmol)二氯甲烷(20ml),室溫攪拌24h,加水和乙醚萃取,然后再用乙醚萃取兩次,合并有機相用水洗一次,再用飽和食鹽水洗一次,用無水硫酸鈉干燥有機相,過濾后減壓蒸出有機溶劑,并用石油醚柱層析得到淡黃色油狀液體化合物11(1.01g,收率77%)
在干燥的并持續通入氮氣的反應器中下加入化合物11(0.92g,1.4mmol)、干燥的四氫呋喃(20ml)。降溫至-78℃,然后向溶液中緩慢滴加1.6M的t-BuLi(2.6ml,4.2mmol)。反應溶液在-78℃下攪拌,反應1.5小時。開始滴加DMF(0.323ml,4.2mmol),滴加完畢后使反應液升溫至室溫繼續反應20h,用水萃滅反應后,用乙醚萃取兩次,合并有機相用水洗一次,再用飽和食鹽水洗一次,用無水硫酸鈉干燥有機相,過濾后減壓蒸出有機溶劑,并用石油醚柱層析得到淡黃色油狀液體化合物12(0.47g,收率61%)
在干燥的并持續通入氮氣的反應器中,避光下加入化合物12(0.47g,0.85mmol)、BF3-Et2O(24mg,0.17mmol)、Amberlyst-15(31mg,0.17mmol)、干燥的乙醚(15ml)。室溫反應4h,反應完畢后用乙醚萃取兩次,合并有機相用水洗一次,再用飽和食鹽水洗一次,用無水硫酸鈉干燥有機相,過濾后減壓蒸出有機溶劑,并用石油醚柱層析,然后用氯仿重結晶得到蒼白色晶體化合物C-1(0.17g,收率37%)。
實施例6化合物(C-5)的制備
與實施例5不同之處在于,將3-溴-5-正丁基噻吩-2-甲醛用3-溴-5-正辛基噻吩-2-甲醛替換,而后按照實施例5的步驟制備獲得化合物(C-5),如下式所示。
將上述實施例獲得的化合物在常規溶劑的溶解度見表1所示。
表1有機半導體化合物在常規有機溶劑的溶解度
a溶解度是在1毫升溶劑中所能溶解的化合物的質量。
實施例7~實施例12硅片基板有機薄膜晶體管的制備
以重摻雜的n型硅片為基板;
所述基板上覆蓋有厚度為300nm、設置有柵極的二氧化硅介電層,所述柵極為重摻雜的n型硅片;
所述二氧化硅介電層采用辛基三氯化硅烷修飾,形成修飾層;
所述修飾層上覆蓋有厚度在30納米~60納米的半導體層,所述半導體層的制備過程分別如下:選用實施例1、實施例2、實施例3、實施例4、實施例5、實施例6得到的可溶性并噻吩衍生物作為半導體材料,分別配成濃度為0.5wt%的甲苯溶液,在轉速為1000rpm、旋轉時間為60秒的條件下形成薄膜,然后進行退火,所述退火的溫度和時間參見表2,表2為本發明實施例提供的有機薄膜晶體管的主要工藝參數和性能。退火后,再沉積厚度為50納米的金作為源電極和漏電極,形成的導電溝道的寬長比為30(W/L=3000微米/100微米=30),得到半導體層,最后形成有機薄膜晶體管。得到有機薄膜晶體管后,而后采用上述獲得化合物按照常規方式分別測定其轉移曲線,各載流子遷移率、開關電流比的結果參見表2。
表2本發明實施例13~17提供的有機薄膜晶體管的工藝參數和性能
由表2可知,采用本發明實施例5提供的可溶性并苯噻吩衍生物制備有機薄膜晶體管,其載流子遷移率可達到0.62cm2V-1s-1,開關電流比達106。
由以上實施例可知,采用本發明提供的可溶性并苯噻吩衍生物能夠獲得具有較高遷移率的有機薄膜晶體管。本發明不限于上述實施例。一般來說,本發明所公開的有機薄膜晶體管可加工形成二維和三維集成器件中的元件。這些集成器件能應用于柔性集成電路、有源矩陣顯示等方面。使用基于本發明的有機薄膜晶體管元件可以低溫溶液加工。
以上實施例的說明只是用于幫助理解本發明的方法和核心思想。應當指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以對本發明進行若干改進和修飾,這些改進和修飾也落入本發明權利要求的保護范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 苯并雙(噻二唑)衍生物、包含其的油墨、以及使用了其的有機電子裝置的制造方法
- 具有含硫取代基的殺有害生物活性雜環衍生物的制造方法與工藝
- 具有含硫取代基的殺有害生物活性雜環衍生物的制造方法與工藝
- 一種苯并噻咯類衍生物及其制備方法和有機發光器件與制造工藝
- 苯并噻吩并咔唑類衍生物及其制備方法和有機發光器件與制造工藝
- 苯并二氮雜*衍生物的2-萘磺酸鹽及晶型和它們的制備方法與制造工藝
- 靛紅雜合喹唑啉類化合物合成的靛紅衍生物及其在制備抗腫瘤藥物中的應用的制造方法與工藝
- 一種雜環衍生物及使用該雜環衍生物的有機發光器件的制造方法與工藝
- 一種二氰基甲烯基苯并四氫呋喃衍生物及其制備方法與制造工藝
- 一種含氮雜環衍生物及使用該含氮雜環衍生物的有機發光器件的制造方法與工藝
- 苯并二氮雜*衍生物的2-萘磺酸鹽及晶型和它們的制備方法與制造工藝
- 一種二氰基甲烯基苯并四氫呋喃衍生物及其制備方法與制造工藝
- 苯并吲唑類衍生物及其制備方法、應用與制造工藝
- 一種二苯并環庚烷衍生物的制備方法與制造工藝
- Atropurpuran的苯并咪唑基和嗎啉基衍生物組合物用于防治胰腺纖維化的制造方法與工藝
- 一種苯并咪唑衍生物鎘配合物及其制備方法和應用
- 一種咪唑并吡啶巰乙酸類衍生物及其制備方法與應用
- Artalbic acid的O?(苯并咪唑基)乙基與O?(二羥乙胺基)乙基衍生物的組合物在抗病毒 ...的制作方法
- 阿掏比克酸苯并咪唑基與二羥乙胺基衍生物的組合物用于制備抗骨質疏松藥物的制作方法
- 阿掏比克酸苯并咪唑基與二羥乙胺基衍生物的組合物用于制備防治胰腺纖維化藥物的制作方法
- 6,7,8,9?四氫?5H?苯并[7]輪烯?2?烷基胺類化合物及其用途的制造方法與工藝
- 用于PHOLED的具有氮雜?二苯并噻吩或氮雜?二苯并呋喃核的新材料的制造方法與工藝
- 多孔ZSM?5沸石與γ?Al2O3復合材料的合成及制備加氫脫硫催化劑的制造方法與工藝
- 一種含有氟噻蟲砜和五氟蟲腙的殺蟲組合物的制造方法與工藝
- 苯并雙(噻二唑)衍生物、包含其的油墨、以及使用了其的有機電子裝置的制造方法
- 苯并噻二唑胺的制造方法與工藝
- 用于有機電致發光器件的材料的制造方法與工藝
- 用于有機電致發光器件的材料的制造方法與工藝
- 一種苯并噻咯類衍生物及其制備方法和有機發光器件與制造工藝
- 苯并噻吩并咔唑類衍生物及其制備方法和有機發光器件與制造工藝
- 一種噻吩并[3,2?d]嘧啶類HIV?1逆轉錄酶抑制劑及其制備方法和應用與流程
- 一類噻吩稠合苯并噁二唑衍生物及其合成方法與流程
- 一種可溶性苯并噻吩衍生物及其制備和應用的制作方法與工藝
- 含噻吩磺酰胺結構的苯并喹唑啉類酪氨酸激酶抑制劑、制備方法及用途與流程
- 一種含噻吩磺酰胺結構的乙氧苯并喹唑啉類酪氨酸激酶抑制劑、制備方法及用途與流程
- 一類含噻吩磺酰胺結構的苯并喹唑啉類酪氨酸激酶抑制劑及其用途的制作方法與工藝
- 一種含噻吩磺酰胺結構的乙氧苯并喹唑啉類酪氨酸激酶抑制劑及用途的制作方法與工藝
- 一種基于S,S?二氧二苯并噻吩衍生物的給受體型交替共軛聚合物發光材料及其制備方法與應用與流程
- 一種三苯基硅基相連苯并噻吩衍生物及使用該衍生物的有機發光器件的制作方法與工藝
- 苯深度脫噻吩的鎳基吸附劑及其制備方法和應用與流程