N末端截短的白介素?38的制作方法
本發明涉及N-末端截短的白介素(IL)-38蛋白、或其功能變體,還涉及編碼該截短的IL-38肽的核酸和載體以及包含這些核酸或載體的重組細胞。本發明顯示,IL-38經過N末端加工,并且該細胞因子的截短形式在巨噬細胞中起免疫活化拮抗劑的作用。這表明了該截短的細胞因子在治療和預防自身免疫病癥中的用途。本發明還提供了包含該截短的IL-38蛋白的藥物組合物,以及用于篩選截短的IL-38的功能的調節劑的方法。
描述
IL-1家族細胞因子和受體為尤其調節免疫的異質性的蛋白組。最初,4種IL-1家族細胞因子被集中地表征(IL-lα、IL-1β、IL-lRa和IL-18),揭示了免疫調節的基本原理,其中的一些已轉化為臨床應用。剩下的7種IL-1家族細胞因子是通過基因數據庫的軟件分析(silico analysis)而鑒定(IL-33、IL-36α、IL-36β、IL-36γ、IL-36Ra、IL-37和IL-38)。近些年,進行了一些重要研究來考察它們誘導或調節免疫應答的相關性。結論是,IL-1家族細胞因子在免疫方面展示了廣譜功能,包括誘導Th1和Th2炎癥以及調節抗炎或前消散效應。在機制水平上,觸發炎癥是通過具有受體激動劑(IL-lα、IL-1β、IL-18、IL-33、IL-36)功能的IL-1家族細胞因子所介導,這通過IL-1家族受體拮抗劑(IL-lRa,IL-36Ra)來抗衡。值得注意的是,完全的受體激動劑或拮抗劑功能一般要求IL-1家族細胞因子的N末端加工,通常是從前體生成成熟的細胞因子。這些事件中最突出的地方可能是利用炎性體的IL-1β成熟。
IL-1家族受體以存在細胞外免疫球蛋白結構域和信號轉導所需的細胞內TIR結構域為特征。IL-1受體家族包括具有已知配體和功能的4個成員:IL-1R1(IL-1RI)、IL-1R4(ST2)、IL-1R5(IL-18R)、IL-1R6(IL-lRrp2);2個共受體:IL-1R3(IL-lRAcP)、IL-1R7(IL-18AcP);1個誘騙受體:IL-1R2(IL-IRII);以及3個孤兒受體TIR8(SIGIRR)、TIGIRR-1(IL-1RAPL2)、TIGIRR-2(IL-1RAPL1)。孤兒受體的命名仍是不明確的。IL-1RAPL1也稱為TIGIRR-2,起初命名為IL-1R8。但在最近,其被稱作IL-1R9或IL-1R10。為了避免混淆,在當前手稿中,發明人將使用術語IL-1RAPL1。IL-1RAPL1在腦中高度表達,參與了小腦發育、智力遲鈍和認知缺陷。與IL-1受體家族其它成員的主要結構差異為IL-1RAPL1的細胞內結構域中C末端150個氨基酸長的延伸,這也存在于其緊密同源物IL-1RAPL2和調節性受體TIR8中。這種結構在細胞內信號轉導中的作用還未有描述。功能性研究提示,IL-1RAPL1的活化和下游信號轉導機制與IL-1受體家族的其它成員不同。有證據表明,IL-1RAPL1選擇性地激活JNK,而JNK參與了免疫活化等。實際上,功能RNAi篩選揭示,IL-lRAPLl調節與凋亡細胞相互作用后的巨噬細胞表型。
IL-38,也稱為IL1F10,為IL-1家族的最新增加。其與IL-1Ra共有41%的同源性,與IL-36Ra有43%的同源性,因此被認為是IL-1受體拮抗劑。實際上,最近顯示,IL-38可結合IL-1R6,由此減少人白色念珠菌(C.albicans)刺激后或以低濃度施用添加IL-36后的細胞因子生成。然而,注意到LPS刺激聯合IL-38后所生成的細胞因子的增加。通常,IL-38多態性與出現自身炎性病理學(例如脊椎關節炎、類風濕性關節炎或牛皮癬性關節炎)的增加的易感性相關,或者與CRP水平相關,提示了IL-38在調節引起這些狀況的機制中的作用。
細胞因子包括大量的哺乳動物免疫調節激素,它們由免疫系統的細胞分泌。它們通過與細胞表面的特異受體相互作用來發揮它們的生物學效應。因此,對細胞因子的生物學應答是由細胞因子的存在和其受體分子的表達二者來調節的。包括自身免疫、炎癥和癌癥疾病在內的很多哺乳動物疾病是與增加或以其它方式改動的細胞因子或細胞因子受體水平相關的,它們可能促進了免疫系統調節紊亂和疾病進展。因而,就這些疾病狀態而言,能夠阻斷細胞因子的免疫調節或炎癥效應的化合物應該具有明顯的治療活性。
產生與不希望的免疫應答相關的免疫抑制的常規策略是基于廣譜免疫抑制藥物。此外,為了維持免疫抑制,免疫抑制藥物治療通常是終生命題。不幸的是,廣譜免疫抑制藥的使用與嚴重副作用(例如腫瘤、感染、腎毒性和代謝病癥)的風險相關聯。于是,新的免疫抑制療法可能是有益的。
所以,直至今天,沒有令人滿意的治療手段來治療或預防自身免疫疾病,并且始終亟需另外的免疫抑制劑,以便推進對患有與病理學或者甚至長期活化的免疫系統相關的疾病的患者的醫療護理。
在第一方面,以上問題是通過分離的截短的IL-38蛋白、或其功能變體來解決,其中所述截短的IL-38蛋白與SEQ ID NO:1的氨基酸序列比較為N末端截短的。
在本文中,術語“截短的IL-38蛋白”指這樣的IL-38多肽,其中已從全長IL-38多肽的氨基末端(或N-末端)區域去除氨基酸殘基。在本發明上下文中,“截短的IL-38蛋白”不包括如SEQ ID NO:1中顯示的全長序列。
術語蛋白的“功能變體”在本文是指這樣的變體蛋白,其中與本發明相關的定義為親和性和穩定性的功能基本上是保留的。因此,與所述功能無關的一個或多個氨基酸可以被替換。術語“功能變體”還應理解為指來自其它哺乳動物的同源物。優選地,本發明的功能變體保留了本文描述如SEQ ID NO:2顯示的截短的IL-38蛋白的免疫抑制能力。
在優選的實施方案中,所述分離的截短的IL-38蛋白或其功能變體包括與SEQ ID NO:1的氨基酸序列(全長IL-38)有至少50%序列相同性的氨基酸序列,特征在于該截短的IL-38蛋白不包括SEQ ID NO:1中所示的全長氨基酸序列。
術語“序列相同性”(或“序列同源性”)表明了對兩等長的氨基酸序列或兩等長的核苷酸序列之間的相同性程度的定量衡量。如果兩對待比較序列不是等長的,則它們必須排列為最佳可能的匹配,帶有空位插入或者選擇性地在蛋白序列的末端截短。
更優選的最小序列相同性百分比為至少70%,例如至少80%、至少85%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、以及至少99.5%、最優選100%(與SEQ ID NO:1中顯示的序列相比),條件是所述截短的IL-38序列不包括如SEQ ID NO:1中所示的IL-38全長序列。
在優選的實施方案中,與野生型IL-38蛋白(SEQ ID NO:1)比較,本發明所述截短的IL-38蛋白在其N末端截短了2至50個氨基酸。優選地,與SEQ ID NO:1中所示的蛋白比較,所述截短的IL-38蛋白在其N末端截短了9、10、11、12、13、14、15、16、17、18、19或20個氨基酸。最優選為19個。
根據本文描述的發明,N末端截短優選包含SEQ ID NO:1的1至30位之間的至少2個、優選5個、最優選10個、最優選20個相鄰的氨基酸。
在其它實施方案中,本發明的截短的IL-38蛋白具有不同于SEQ ID NO:1的前100、50、30、20個氨基酸的N末端。
在其它實施方案中,本發明的截短的IL-38蛋白具有這樣的N末端,其中該N末端的前50個氨基酸不同于SEQ ID NO:1的前50個氨基酸。
在其它實施方案中,本發明的截短的IL-38蛋白具有這樣的N末端,其中該N末端的前20個氨基酸不同于SEQ ID NO:1的前20個氨基酸。
作為選擇,本發明的截短的IL-38蛋白不包含與SEQ ID NO:1的1至20位之間的序列至少80%相同的序列。
進一步最優選的是由與SEQ ID NO:2(20-152_IL-38)有至少80%、85%、90%、95%或100%序列相同性的氨基酸序列組成的截短的IL-38蛋白。如果通過重組表達本發明的截短的IL-38蛋白來生產,則該截短的IL-38蛋白的序列特征在于在N末端存在額外的甲硫氨酸。這些蛋白可以在表達后接受純化過程,以獲得本發明的分離的截短的IL-38蛋白。
“分離的”和“純化的”指與其天然環境分開,并且擺脫了約60%至約99%、優選80%至99%的與其天然結合的其它組分的任何分子或化合物。
因此,在這方面,尤其優選的本發明的截短的IL-38蛋白在1位包含N末端甲硫氨酸;這些蛋白為純粹人工的,非天然存在。
在本文中,術語“重組的”指重組或者合成產生的蛋白或核酸構建體,例如,對于蛋白的情況,通過在宿主細胞中翻譯特定載體或質粒相關的系列指定的核酸元件或表達盒的RNA轉錄本。在本文中,術語“重組的”不包括天然發生的事件(例如,自發突變、天然轉化/轉導/轉座)引起的細胞或載體變動,例如沒有刻意的人類介入而發生的那些情況。
“宿主細胞”指這樣的細胞,其含有載體或表達盒,并支持它們的復制和/或表達。宿主細胞可以為原核細胞,例如大腸桿菌(E.coli),或真核細胞,例如酵母、昆蟲、兩棲動物、植物細胞和哺乳動物細胞。優選地,宿主細胞為細菌或原核細胞。
在本文中,“載體”包括對用在宿主細胞轉染中并且其中可插入多核苷酸的核酸的指代。載體一般是復制子。
在本文中,術語“蛋白(protein)”或“蛋白(proteins)”指多肽或其任何部分。
術語“多肽”指氨基酸的聚合物,并不是特定長度的產物;因此,肽、寡肽、和蛋白都包括在多肽的定義中。該術語也并不是多肽的表達后修飾或者排除多肽的表達后修飾,例如糖基化、乙酰化、磷酸化等等。包括在該定義內的例如有:含有一個或多個氨基酸類似物(包括,例如非天然氨基酸等)的多肽,具有取代鍵的多肽,以及本領域中已知的修飾,無論是天然發生的還是非天然發生的。
在本文中,術語“重組蛋白”是指(1)半合成的或合成起源的多肽,源于采用重組DNA技術連接在一起的不同來源的重組DNA分子的表達;(2)半合成的或合成起源的多肽,由于其來源或者操作,其不與天然結合的蛋白的全部或一部分結合;(3)半合成的或合成起源的多肽,其連接至一多肽,該多肽不同于其天然連接的多肽;或(4)半合成的或合成起源的多肽,其不天然出現。
本發明還涵蓋化學上或以其它方式修飾的截短的IL-38蛋白。本文描述的多肽的修飾形式例如為經翻譯后修飾的IL-38蛋白。翻譯后修飾可以為所表達蛋白的糖基化。
上文描述的現有技術的問題在其一另外方面通過提供包含編碼(coding或encoding)本文描述的截短的IL-38蛋白的序列的核酸來解決。術語“編碼”(coding或encoding)是指核苷酸序列編碼一個或多個氨基酸的能力。該術語不需要起始或終止密碼子。氨基酸序列可編碼在多核苷酸序列和其互補物提供的6種不同閱讀框的任何一種中。
本發明的核酸可以包含這樣的序列,當表達時生成由本發明的截短的IL-38蛋白組成的多肽,但其不為SEQ ID NO:1的全長IL-38蛋白。
本發明的另一方面還涉及包含上文描述的核酸的載體。最優選的是,本發明的載體為表達載體。
“表達載體”為重組或合成產生的核酸構建體或序列,具有特定的核酸元件,使得能在宿主細胞中轉錄和/或表達另一核酸。表達載體可為質粒、病毒、或核酸片段的一部分。在一個實例中,表達載體為DNA載體,例如質粒,其包含至少一個啟動子序列和至少一個終止子序列(例如,多聚腺苷酸化序列),以及任選的復制起點(ori)序列,和任選的篩選或可篩選標記序列。任選地,該表達載體可以還包含至少一個感興趣的核苷酸編碼序列,其編碼至少一種多肽,其中所述至少一個啟動子序列與該至少一個編碼序列可操作地連接。術語“表達”包括參與多肽生成的任何步驟,包括但不限于,轉錄、轉錄后修飾、翻譯、翻譯后修飾、和/或分泌。
本文還提供了包含本文描述的核酸或載體或表達載體的重組細胞。本發明的重組細胞優選不為人胚胎干細胞。本發明的優選重組細胞例如為細菌細胞,例如大腸桿菌或其它表達系統,或也可為動物細胞,例如昆蟲細胞、哺乳動物細胞和人細胞。
本文描述的化合物和組合物可以用在各個醫療領域,尤其是作為活性治療劑用于在需要治療的受試者中治療或預防以免疫應答病理性激活為特征的疾病。由本發明的化合物治療的優選疾病將在下文描述。
因此,在另外方面,本發明還提供了藥物組合物,其包含本發明前文描述的實施方案的截短的IL-38蛋白、或核酸、載體或重組細胞,用于用在醫藥中,優選用在治療或預防免疫或炎性疾病中。
在本發明上下文中,免疫或炎性疾病優選選自自身免疫疾病,例如感染性休克,失血性休克,關節炎,例如脊椎關節炎,類風濕性關節炎,牛皮癬性關節炎或骨關節炎,炎性腸病,多發性硬化和代謝疾病如動脈硬化和I型糖尿病。其它的適應癥包括對IL-1β的抑制劑治療有應答的疾病,例如Muckle-Wells綜合征、CAPS(cryopyrin相關周期性發熱綜合征)、家族性地中海熱、斯蒂爾病、白塞病和糖尿病。
關節炎包括骨關節炎,類風濕性關節炎作為損傷或晶體沉積結果的關節炎性關節等為常見的炎性狀況,它們將受益于抗炎蛋白的治療應用,例如本發明的截短的IL-38蛋白。例如,類風濕性關節炎(RA)為影響整個身體的全身性疾病,為最常見形式的關節炎之一。其特征在于襯在關節下的隔膜的炎癥,其引起疼痛、僵直、升溫、發紅和腫脹。炎性細胞釋放可以消化骨骼和軟骨的酶。作為類風濕關節炎的結果,發炎的關節襯里(滑膜)可侵入并損傷骨骼和軟骨,引起關節退化和嚴重疼痛以及其它生理效應。受累關節可能喪失其形狀和排列,導致疼痛和喪失運動能力。
類風濕性關節炎(RA)為免疫介導疾病,尤其以炎癥和隨后的組織損傷為特征,導致嚴重殘疾和增加的死亡率。各種細胞因子在類風濕關節內局部生成。眾多的研究已證實,兩種原型促炎細胞因子IL-1和TNF-α在涉及滑膜炎癥和進行性關節破壞的機制中起重要作用。實際上,在患有RA的患者中以TNF-α和IL-1抑制劑施用已導致炎癥的臨床和生物學跡象的明顯改善和骨骼侵蝕和軟骨破壞的放射跡象的減弱。但是,盡管有這些令人鼓舞的結果,有相當百分比的患者對這些藥物無應答,提示其它的調節物也參與了關節炎的病理生理學(Gabay,Expert.Opin.Biol.Ther.2(2):135-149.2002;Astry,J.Interferon Cytokine Res.31(12):927-40.2011)。
對于藥物應用,本發明的截短的IL-38蛋白被配制為用于常規方法的胃腸外遞送,特別是靜脈內或者皮下遞送。靜脈內施用將通過團注、控釋(例如,采用微型泵或其它適合的技術)、或通過在一個至數個小時的典型時間段內輸注來進行。通常,藥物制劑將包括與藥學上可接受的載體(例如,鹽水,緩沖鹽水,5%葡萄糖水溶液等)聯合的造血蛋白。制劑還可以包括一種或多種賦形劑、防腐劑、增溶劑、緩沖劑、防止蛋白在小瓶表面損失的白蛋白,等等。當采用這種聯合治療時,細胞因子可以組合在單一制劑中,或者可以在不同的制劑中施用。配制方法為本領域所公知的,例如在文獻(Remington's Pharmaceutical Sciences.Gennaro,ed.,Mack Publishing Co.,Easton PA,1990)中有公開,通過參考將其并入本文。
治療劑量通常在每天0.1至100mg/kg患者體重的范圍內,優選每天0.5-20mg/kg,精確劑量由臨床醫師根據公認的標準,并考慮待治療的狀況的性質和嚴重性、患者特征等來確定。劑量確定處于本領域普通技術人員的能力范圍內。所述蛋白一般在不超過28天的時間內施用。更常見地,所述蛋白在一周或更短時間內施用,經常在1至3天的時間內施用。通常,治療上有效量的本發明的截短的IL-38蛋白為足以產生病理性炎癥反應的臨床顯著降低的量。
通常,施用的截短的IL-38蛋白的劑量將隨諸如患者年齡、體重、身高、性別、一般醫學狀況和之前的病史的因素而變化。典型地,希望向接受者提供約1pg/kg至10mg/kg(藥物量/患者體重)范圍的截短的IL-38蛋白劑量,不過根據情況不同也可以以更低或更高劑量施用。在下文提供本發明藥物組合物的具體實施方案。
在另外的方面,本發明提供了在需要治療的受試者中治療或預防病理性炎性疾病的方法。本發明的方法可以包括向所述受試者施用治療有效量的本文描述的化合物或本發明的組合物的任一種的步驟。
本發明的另一方面涉及調節細胞的免疫應答的方法,該方法包括讓所述細胞與本發明的截短的IL-38蛋白接觸,或者通過在所述細胞中表達本發明的核酸。
在優選的實施方案中,該方法為離體或體外方法。
“調節免疫應答”可以為對JNK信號轉導的抑制,尤其是抑制IL-6釋放和TH17生成。可以通過在所述細胞中使用JNK報告基因構建體來觀察對JNK信號轉導的抑制。一種廣泛使用的報告基因為AP-1啟動子驅動的報告基因。
用在本發明所描述的方法中的細胞優選為哺乳動物細胞,最優選人細胞。還優選該細胞為免疫細胞,最優選白細胞,進一步更優選巨噬細胞。
本發明的另一方面涉及篩選截短的IL-38的活性的調節劑的方法,包括以下步驟:
a.提供細胞,
b.讓所述細胞與微生物相關分子模式(MAMP)、病原體相關分子模式(PAMP)、或凋亡細胞上清液(ACM)接觸,
c.讓所述細胞再與本發明的截短的IL-38蛋白和候選調節劑接觸,
d.確定所述細胞中的JNK激活,
其中,所述細胞中JNK激活與對照細胞或參考值相比的增加,表明該候選調節劑為截短的IL-38的拮抗劑,JNK激活與對照細胞或參考值相比的降低,表明該候選調節劑為截短的IL-38的激動劑。
根據本發明的PAMP或MAMP可以選自細菌脂多糖(LPS)、細菌鞭毛蛋白、來自革蘭氏陽性細菌的脂磷壁酸、肽聚糖、以及通常與病毒相關的核酸變體,例如雙鏈RNA(dsRNA),或未甲基化的CpG基序。
用在本發明的篩選方法中的所述細胞優選在細胞表面表達截短的IL-38的受體,例如通過在所述細胞中異位表達IL-1RAPL1。
候選調節劑優選小分子,小核酸,例如小RNA,或蛋白,例如抗體。
所述JNK激活優選通過AP-1報告基因構建體來確定。這種報告基因構建體可以為基于熒光素酶的、基于酶的或基于熒光蛋白的。也可以例如通過定量PCR(qPCR)來確定直接的JNK靶基因表達。
本發明還提供了通過本文描述的方法鑒定的截短的IL-38的活性調節劑。
疾病和狀況
本發明提供了截短的IL-38蛋白,其可在治療或預防各種疾病中用作治療劑。根據本發明,這些化合物和組合物尤其可用于治療和/或預防以病理性激活的免疫或炎性應答為特征的狀況。具體地,本發明尋求提供對以細胞因子的病理學活性為特征的狀況的治療,這些細胞因子例如為白介素-6和白介素-17,它們已被證實參與了多系列的慢性炎性和自身免疫病癥。
在本發明的上下文中,自身免疫疾病為自身免疫應答導致的病癥。自身免疫疾病為對自身抗原的不恰當的和過度的應答的結果,或者為例如經由細胞因子的免疫應答信號轉導的病理性激活的結果。自身免疫疾病的實例包括但不限于,阿狄森氏病、斑禿、強直性脊椎炎、自身免疫性肝炎、自身免疫性腮腺炎、克羅恩病、糖尿病(I型)、營養不良型大皰性表皮松解癥、附睪炎、血管球性腎炎、格式(Graves)病、巴雷(Guillain-Barr)綜合征、橋本病、溶血性貧血、系統性紅斑狼瘡、多發性硬化、重癥肌無力、尋常型天皰瘡、牛皮癬、風濕熱、類風濕性關節炎、結節病、硬皮病、斯耶格倫氏綜合征、脊柱關節病、甲狀腺炎、脈管炎、白癜風、粘液水腫、惡性貧血、潰瘍性結腸炎等等。
治療或預防自身免疫疾病的組合物和試劑盒
本申請的另一方面涉及用于治療或預防自身免疫或炎性疾病的組合物和試劑盒。在一個實施方案中,該組合物包含本文描述的諸如蛋白、核酸或重組細胞的化合物,任選地聯合藥學上可接受的載體。
在本文中,用語“藥學上可接受的載體”旨在包括與藥物管理相符的任何和所有的溶劑、增溶劑、填充劑、穩定劑、粘結劑、吸附劑、堿、緩沖劑、潤滑劑、控釋載體、稀釋劑、乳化劑、濕潤劑、潤滑劑、分散介質、涂層、抗細菌或抗真菌劑、等張和吸收延緩劑,等等。將這些介質和試劑用于藥學上有活性的物質在本領域內是公知的。除了任何常規媒介物或試劑與該活性化合物不相容之外,考慮將其用在該組合物中。補充劑也可摻入到該組合物中。在某些實施方案中,該藥學上可接受的載體包括血清白蛋白。
本發明的藥物組合物配制為與其預期的施用途徑相容。施用途徑的實例包括胃腸外施用,例如鞘內、動脈內、靜脈內、皮內、皮下施用,口服,透皮(局部)和跨粘膜施用。
用于胃腸外、皮內或皮下應用的溶液或懸浮液可包括以下組分:無菌稀釋劑如注射用水,鹽水溶液,不揮發油,聚乙二醇,甘油,丙二醇或其它合成溶劑;抗細菌劑例如苯甲醇或對羥基苯甲酸甲酯;抗氧化劑例如抗壞血酸或硫酸氫鈉;螯合劑例如乙二胺四乙酸;緩沖劑例如乙酸鹽,檸檬酸鹽或磷酸鹽以及用于調節張力的試劑如氯化鈉或右旋糖。可用酸或堿,例如鹽酸或氫氧化鈉調節pH。胃腸外配制物可封在安瓿瓶、一次性注射器或多重劑量小瓶中,它們由玻璃或塑料制成。
適合于注射使用的藥物組合物包括無菌水溶液(當水溶時)或懸浮液以及用于臨時制備無菌可注射溶液或懸浮液的無菌粉末。對于靜脈內施用,適合的載體包括生理鹽水、抑菌水、商品名為Cremophor ELTM的物質(BASF,Parsippany,N.J.)或磷酸鹽緩沖鹽水(PBS)。所有情況下,可注射組合物應為無菌的,并且應為流體,其程度為存在易注射性。其在生產和保存條件下必須是穩定的,并且必須能免于微生物如細菌和真菌的污染作用。載體可為溶劑或分散介質,含有例如水、乙醇、多元醇(例如,甘油、丙二醇、以及液體聚乙二醇等)、或它們的適當混合物。例如通過使用諸如卵磷脂的涂層、對于懸浮液的情況通過維持所需的粒徑以及通過使用表面活性劑可維持適當的流動性。預防微生物作用可通過各種抗細菌和抗真菌劑來實現,例如,對羥基苯甲酸酯、氯丁醇、酚、抗壞血酸、硫柳汞,等。很多情況下,優選在組合物中包括等張劑,例如,蔗糖、多元醇如甘露醇、山梨醇和氯化鈉。可注射組合物的延時吸收可通過在組合物中加入延緩吸收的試劑來實現,例如單硬脂酸鋁和明膠。
可通過將活性化合物(例如,神經調節蛋白)以所需量與以上列出的成分的一種或組合摻入適合的溶劑中,然后無菌過濾(需要的話),來制備無菌可注射溶液。通常,通過將活性化合物摻入含有基礎分散介質和所需的來自以上列出成分的其它成分的無菌載體中來制備分散體。對于用于制備無菌可注射溶液的無菌粉末的情況,優選的制備方法為真空干燥和冷凍干燥,其產生活性成分加上任何額外所需成分(來自其之前無菌過濾的溶液)的粉末。
口服組合物通常包括惰性稀釋劑或可食用載體。它們可包封在明膠膠囊中或壓縮為片劑。出于口服治療施用的目的,活性化合物可與賦形劑一起摻混,并以片劑、含片、或膠囊的形式使用。口服組合物還可采用流體載體來制備,以用作漱口水,其中流體載體中的化合物口服施用,在口中來回,并且吐出或咽下。藥學上相容的粘結劑和/或佐劑材料可作為組合物的一部分包括進來。片劑、小丸、膠囊、含片等可含有以下成分或具有類似性質的化合物的任何一種:粘結劑如微晶纖維素、黃蓍膠或明膠;賦形劑如淀粉或乳糖;崩解劑如褐藻酸、商品名為Primogel的物質、或玉米淀粉;潤滑劑如硬脂酸鎂或商品名為Stertes的物質;助流劑如膠體二氧化硅;甜味劑如蔗糖或糖精;或調味劑如薄荷、水楊酸甲酯、或橙香精。
對于通過吸入施用,化合物是以氣溶膠噴霧的形式從含有適合的推進劑(例如氣體形式,諸如二氧化碳)的壓力容器或分配器或噴霧器遞送。
全身施用也可通過跨粘膜或透皮方式進行。對于跨粘膜或透皮施用,將適合于待滲透屏障的滲透劑用在制劑中。這些滲透劑通常在本領域是已知的,包括例如用于跨粘膜施用的洗滌劑、膽汁鹽、以及梭鏈孢酸衍生物。跨粘膜施用可通過使用鼻噴劑或者栓劑來完成。對于透皮施用,藥物組合物配制為軟膏、藥膏、凝膠、或乳膏,如本領域所公知的。
在某些實施方案中,藥物組合物配制為活性成分的持續或控制釋放。可使用生物可降解的、生物相容的聚合物,例如乙烯醋酸乙烯酯、聚酸酐、聚乙醇酸、膠原、聚原酸酯、以及聚乳酸。用于制備這些制劑的方法對本領域技術人員是顯而易見的。這些材料也可在商業上從例如阿爾扎公司(Alza Corporation)和諾華制藥公司(Nova Pharmaceuticals,Inc.)獲得。脂質體懸浮液(包括帶有針對病毒抗原的單克隆抗體的靶向感染細胞的脂質體)也可用作藥學上可接受的載體。這些可按照本領域技術人員已知的方法來制備。
特別有利的是配制單位劑量形式的口服或胃腸外組合物,以方便施用和劑量一致性。單位劑量形式在本文中包括物理上分開的單位,適合作為單一劑量用于待治療的受試者;每個單位含有預定量的活性化合物,經計算與所需的藥物載體聯合會產生期望的治療效果。本發明單位劑量形式的規格取決于并直接依賴于活性化合物的獨特特征和待獲得的具體治療效果以及本領域中將這種活性化合物配制為用于個體治療的內在限制。
這些化合物的毒性和治療功效可通過標準藥學程序在細胞培養物或試驗動物中確定,例如用于確定LD50(50%群體致死劑量)和ED50(50%群體治療有效劑量)。毒性和治療效果的劑量比例為治療指數,其可表示為比例LD50/ED50。顯示出大治療指數的化合物是優選的。盡管可以使用顯示出毒副作用的化合物,但應考慮設計將這些化合物靶向受累組織位點的遞送系統,以便對非感染細胞的潛在損傷降至最小,并由此減小副作用。
從細胞培養物試驗和動物研究獲得的數據可用在制定用于人的劑量范圍中。這些化合物的劑量優選處于包括ED50的循環濃度的范圍內,具有微小毒性或沒有毒性。該劑量可以在該范圍內隨采用的劑型和使用的施用途徑而變化。對于用在本發明方法中的任何化合物,治療有效劑量可從細胞培養測定初步估計。可在動物模型中制定獲得包括在細胞培養中確定的IC50(即,測試化合物實現癥狀的最大半數抑制的濃度)的循環血漿濃度范圍的劑量。這種信息可用于更精確地確定人類可用劑量。藥物組合物可包括在容器、包裝盒、或分散器中,連同施用說明書。
下面將參照附圖和序列,在以下實施例中進一步描述本發明,然而并不限于此。出于本發明的目的,本文提到的所有參考文獻都通過引用將它們的全文并入。在附圖中:
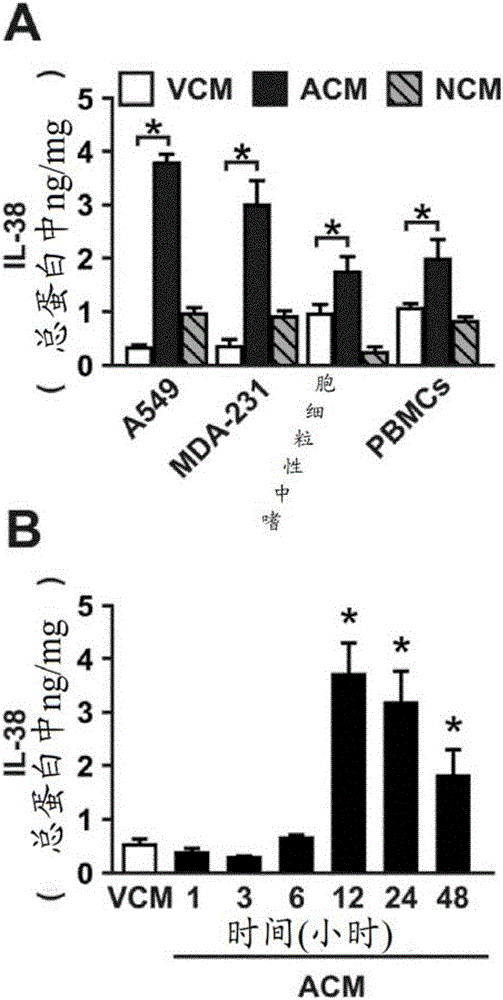
圖1:IL-38是從凋亡細胞分泌的。(A)A549人肺癌細胞,MDA-231乳腺癌細胞、人原代PBMC和人原代嗜中性粒細胞保持存活、經TNF-α(20ng/ml)/CHX(10μΜ)處理以誘導凋亡或者在60℃溫育30min以誘導壞死。收獲存活(VCM)、凋亡(ACM)或壞死(NCM)細胞各自的上清液,并通過ELISA分析IL-38水平。數值為均值±SEM,n=5。(B)在指定的時間通過ELISA分析凋亡的A549細胞的IL-38分泌。數值為均值±SEM,n=5。*p<0.05,ANOVA,有邦弗倫尼(Bonferroni)校正。
圖2:凋亡腫瘤細胞釋放的IL-38調節巨噬細胞中的細胞因子產生。用(A)單獨的LPS(1ng/ml)或聯合重組人IL-38(rhIL-38)10ng/ml或者用(B)單獨的凋亡A549細胞上清液(ACM)或聯合重組人IL-38(rhIL-38)10ng/ml刺激人巨噬細胞。細胞因子的產生采用流式微珠陣列測量,顯示了歸一化的結果。數值為均值±SEM,n=5。(C)用存活的(VCM)或凋亡的A549細胞(ACM)上清液刺激人巨噬細胞,該A549細胞先經非靶向的siRNA(siCtrl)、針對IL-38的siRNA(silL-38)或IL-38過表達載體(oeIL-38)轉染。細胞因子的產生采用流式微珠陣列測量,顯示了歸一化的結果。數值為均值±SEM,n=5。(D)用AP1報告基因構建體轉染人巨噬細胞,并在來自siCtrl和siIL-38A549細胞的ACM刺激24小時后測量熒光素酶活性。從每個實驗值中減去從空白對照轉染細胞獲得的背景測量值。顯示了經歸一化的結果。數據為均值±SEM,n=5。*p<0.05,ANOVA,有邦弗倫尼校正。
圖3:IL-38結合IL-1R6和IL-lRAPLl。(A,B)用LPS或ACM刺激人巨噬細胞6和24h。IL-1R6和IL-lRAPLl的mRNA表達(A)和細胞表面蛋白表達(B)分別通過RT-qPCR和FACS測量。數值為均值±SEM,n=5。(C,D)巨噬細胞為對照(Ctrl)或者與50ng/ml IL-38在冰上溫育15min,然后用抗IL-lR6或抗IL-lRAPLl以及它們各自的PE偶聯第二抗體染色。顯示了代表性的流式細胞直方圖(C)和中值PE強度的統計定量(D)。數值為均值±SEM,n=5。(E)IL-38與固定化的IL-1R6和IL-lRAPLl的結合動力學。用0.5μg的人IL-1R6和IL-lRAPLl細胞外結構域-Fc嵌合體包被96孔板,并與所示的遞增量的人重組IL-38一起溫育。采用生物素化的單克隆IL-38抗體來檢測結合至受體的細胞外結構域的IL-38。數據為均值±SEM,n=5。*p<0.05,ANOVA,有邦弗倫尼校正。
圖4:IL-1R6和IL-lRAPLl在細胞因子產生中的作用。用非靶向siRNA(siCtrl)或針對(A)IL-lRAPLl(silLlRAPLl)或(B)IL-1R6(siILlR6)的siRNA轉染巨噬細胞,并用VCM和ACM刺激24h。細胞因子的產生采用流式微珠陣列測量,顯示了歸一化的結果。數值為均值±SEM,n=5。*p<0.05,ANOVA,有邦弗倫尼校正。
圖5:IL-38調節Th17應答。人T細胞用抗CD3/抗CD28微珠激活,用商品名為eFluor 670的物質進行染色,并用之前經來自對照A549細胞(ACMshCtrl/ΜΦ)或IL-38敲除A549細胞(ACMshIL-38/ΜΦ)的ACM刺激的巨噬細胞的上清液進行刺激。7天后,測量(A)細胞因子的產生和(B,C)細胞增殖。(A)采用流式微珠陣列來定量細胞因子,顯示了歸一化的結果。數值為均值±SEM,n=10。采用商品名為eFluor 670的稀釋液確定T細胞增殖。顯示了(B)所有增殖T細胞的統計定量和(C)代表性流式細胞直方圖。數值為均值±SEM,n=10。*p<0.05,ANOVA,有邦弗倫尼校正。
圖6:IL-38為N末端截短的。(A)顯示了IL-38中的IL-36家族特征性一致基序,這確定了該細胞因子在N末端的推測切割位點。(B)C末端帶myc標簽的IL-38在A549細胞中過表達,然后將其用于ACM生成。在采用抗myc覆蓋的微珠免疫沉淀該過表達的IL-38之后,進行2D凝膠電泳(在pH 4-7等電聚焦,接著是聚丙烯酰胺凝膠分離),并將單克隆抗myc抗體用于檢測經蛋白轉移至硝酸纖維素膜上的免疫沉淀的IL-38。(C)將考馬斯染色的2D凝膠用于挑取推測的IL-38斑點,通過質譜儀對其進行分析。顯示了對不同的IL-38斑點所鑒定的IL-38N末端肽。
圖7:全長和截短的IL-38在細胞因子產生和結合至IL-lRAPLl方面有相反的作用。(A,C)人巨噬細胞(A)未經處理或者(C)先前經非靶向siRNA(siCtrl)或針對IL-lRAPLl(silLlRAPLl)的siRNA轉染,并用單獨的重組人IL-1β50ng/ml或者聯合不同濃度的重組人全長(IL-38aal-152)或剪切的(IL-38aa20-152)IL-38刺激6h。24小時后,上清液中的IL-6濃度采用流式微珠陣列測量,顯示了歸一化的結果。數值為均值±SEM,n=7。(B)全長和剪切的IL-38與固定化的IL-lRAPLl的結合動力學。用0.5μg的人IL-lRAPLl細胞外結構域-Fc嵌合體包被96孔板,并與所示的遞增量的人重組IL-38aal-152或IL-38aa20-152一起溫育。采用生物素化的單克隆IL-38抗體對結合至受體的細胞外結構域的IL-38進行檢測。數據為均值±SEM,n=5。*p<0.05,ANOVA,有邦弗倫尼校正。
圖8:在HEK細胞中由IL-38調節的IL-lRAPLl誘導的信號轉導途徑。用IL-lRAPLl過表達質粒聯合(A,D)AP1、(B,E)NFκB或(C)IL-6報告基因構建體共轉染HEK細胞。將轉染了報告基因構建體以及空質粒而不是IL-1RAPL1過表達質粒的HEK細胞用作對照(空白對照)。(A,B)用IL-1β(50ng/ml)刺激HEK細胞24h,測量AP1或NFκB活性。顯示了經歸一化的結果。數值為均值±SEM,n=15。(C)使用在指出的轉錄結合位點中有點突變的IL-6報告基因構建體。轉染后,將細胞再溫育24h,并測量IL-6啟動子依賴的熒光素酶活性。結果表示為相對于空白對照轉染細胞的誘導倍數。數值為均值±SEM,n=10。(D,E)轉染后,添加新鮮培養基(Ctrl)或不同濃度的IL-38aal-152或IL-38aa20-152,將細胞再溫育24h。結果表示為相對于空白對照轉染細胞的誘導倍數。數值為均值±SEM,n=15。(F)ILIRAPL1在HEK細胞中過表達,并且在轉染后,向細胞添加新鮮培養基或IL-38aal-152或IL-38aa20-152(25ng/ml),接著再溫育24h。進行磷酸化的JNK和p38的細胞內染色并通過FACS測量。結果表示為相對于空白對照轉染細胞的誘導倍數。數值為均值±SEM,n=5。*p<0.05,ANOVA,有邦弗倫尼校正。
圖9:IL-38調節巨噬細胞中的AP1活性。用空載體、AP1、NFκB或IL-6報告基因構建體轉染人巨噬細胞。轉染后,用單獨的IL-1β(50ng/ml)或者聯合IL-38aal-152或IL-38aa20-152(20ng/ml)刺激巨噬細胞24h。測量熒光素酶活性。從每個實驗值中減去從空白對照轉染細胞獲得的背景測量值。顯示了經歸一化的結果。數值為均值±SEM,n=7。*p<0.05,ANOVA,有邦弗倫尼校正。
SEQ ID NO:1
MCSLPMARYYIIKYADQKALYTRDGQLLVGDPVADNCCAEKICILPNRGLARTKVPIFLGIQGGSRCLACVETEEGPSLQLEDVNIEELYKGGEEATRFTFFQSSSGSAFRLEAAAWPGWFLCGPAEPQQPVQLTKESEPSARTKFYFEQSW
SEQ ID NO:2
LYTRDGQLLVGDPVADNCCAEKICILPNRGLARTKVPIFLGIQGGSRCLACVETEEGPSLQLEDVNIEELYKGGEEATRFTFFQSSSGSAFRLEAAAWPGWFLCGPAEPQQPVQLTKESEPSARTKFYFEQSW
實施例
實施例1:IL-38從凋亡細胞釋放
當進行內部ELISA來確定腫瘤細胞系產生的IL-38水平時,發明人注意到對凋亡性細胞死亡的誘導顯著增加了IL-38向上清液中分泌。與存活的A549肺癌或MDA.231乳腺癌細胞上清液(VCM)相比,是凋亡細胞上清液(ACM)而不是壞死細胞上清液(NCM)中含有大約高10倍水平的IL-38(圖1A)。對于原代人嗜中性粒細胞或PBMCs也是如此,盡管凋亡期間IL-38釋放的增加不是一樣強烈(圖1A)。為了分析IL-38分泌動力學,在凋亡誘導后的不同時間點測量了A549上清液中的IL-38濃度。在凋亡誘導后12h觀察到增強的IL-38分泌(圖1B),與A549細胞中凋亡標志物的出現吻合(數據未示出)。
實施例2:IL-38調節ACM刺激后的細胞因子的產生
凋亡細胞衍生的調節劑具有調節包括細胞因子產生在內的吞噬細胞應答的能力(26)。發明人分析了IL-38在一組細胞因子產生中的作用,這些細胞因子在巨噬細胞被LPS激活后或者在與凋亡細胞相互作用后產生(27)。其中,IL-6和IL-8產生由IL-38調節。與單獨LPS相比,向LPS刺激的巨噬細胞添加重組IL-38,增加了IL-6和IL-8的產生。有趣的是,當用單獨的A549細胞ACM或者聯合重組人IL-38刺激人巨噬細胞,觀察到相反的效果(圖2B)。IL-38抑制ACM誘導的IL-6和IL-8從巨噬細胞分泌。由于ACM已經含有IL-38,所以發明人對內源IL-38是否影響巨噬細胞的細胞因子產生感到好奇。為了回答這個問題,在產生ACM前,讓IL-38在A549細胞中過表達或者敲除。實際上,用產生自IL-38過表達的A549細胞的ACM刺激人巨噬細胞會導致減少的IL-6和IL-8分泌,而用IL-38敲除的A549細胞的ACM刺激則產生更高的IL-6和IL-8濃度(圖2C)。在調節細胞因子產生并由IL-1家族調節的主要轉錄因子中有NFκB和AP1。由于NFκB在巨噬細胞與凋亡細胞相互作用后被阻斷(28),所以發明人想了解內源IL-38是否在應答ACM中調節AP1激活。與對照ACM比較,當將IL-38敲除的A549細胞的ACM應用至經AP1熒光素酶報告基因構建體轉染的巨噬細胞時,發明人注意到含有較低水平IL-38的ACM誘導了更明顯的AP1激活(圖2D)。作為結論,內源IL-38限制了應答凋亡細胞上清液的炎性巨噬細胞激活。
實施例3:IL-38拮抗應答ACM中的ILlRAPLl依賴性細胞因子的產生
發明人假設IL-38通過作為受體拮抗劑來抑制ACM誘導的細胞因子產生。由此,發明人分析了IL-1受體家族的候選者與IL-38的結合。已證實IL-38結合IL-1R6(19),并且發明人觀察到孤兒受體IL-lRAPLl調節ACM刺激后巨噬細胞中細胞因子產生(16)。發明人首先采用qPCR在mRNA水平(圖3A)和通過FACS在細胞表面可得性水平(圖3B)上確定了IL1R6和IL-lRAPLl在ACM或LPS刺激后的巨噬細胞中的表達。IL1R6表達通常較低(圖3C),并在ACM或LPS刺激后進一步在mRNA水平上下調,然而在細胞表面表達水平上并不明顯。相反地,IL-lRAPLl表達是豐富的(圖3C),并且在LPS處理后6h以及ACM處理后6h和24h時被進一步誘導,不論是mRNA水平,還是細胞表面(圖3A,B)。此外,細胞表面的IL-lRAPLl表達在LPS刺激后24小時降低。這些實驗提示,IL-lRAPLl至少是作為IL-38起作用的另外候選者。接著,發明人通過進行競爭測定和受體結合測定來分析IL-38是否結合IL-lRAPLl。對于競爭測定,在分析IL-1R6或IL-lRAPLl的表面表達之前讓人巨噬細胞與重組人IL-38一起溫育。基于低水平的IL-1R6表面表達,難以在細胞表面表達中觀察到由IL-38預溫育導致的差異(圖3C,D)。但是,對于IL-lRAPLl,發明人觀察到IL-38與用于FACS染色的抗體競爭(圖3C,D),表明IL-38可以結合IL-lRAPLl。為了驗證這些結果,進行了直接的受體結合測定。用IL-1R6-Fc和IL-lRAPLl-Fc嵌合體包被平板,向孔中添加不同IL-38濃度,并觀察結合的IL-38。如最近所證實的(19),IL-38確實結合IL-1R6(圖3E)。此外,IL-38還結合IL-lRAPLl(圖3E)。這些結果提示,IL-38可能通過結合IL-lRAPLl來調節細胞因子產生,因此發明人想了解IL-lRAPLl在ACM誘導的細胞因子產生中的作用。在人巨噬細胞中進行IL-1R6或IL-lRAPLl的瞬時敲除,并在ACM刺激后測量巨噬細胞培養物上清液中的IL-6和IL-8水平。ACM刺激后的IL-6和IL-8的產生依賴于IL-lRAPLl(圖4A),而在發明人的模型中IL-1R6不參與細胞因子的產生(圖4B)。
實施例4:IL-38調節Th17應答
接下來,發明人通過分析巨噬細胞上清液對T細胞激活的效果,想了解IL-38依賴的抑制巨噬細胞產生細胞因子的下游后果。發明人分離了原代人T細胞,用抗CD3/抗CD28微珠刺激并將它們與巨噬細胞(先前經ACM和IL-38敲除的A549細胞的ACM刺激)的上清液一起反復溫育。在培養7天后,測量T細胞上清液中的IL-10、IL-17和IFN-γ水平。當巨噬細胞經ACM刺激,它們的上清液減少T細胞的IFN-γ和IL-10產生并稍稍升高IL-17水平(圖5A)。然而,當用缺乏IL-38的ACM刺激巨噬細胞時,它們的上清液強烈升高T細胞的IL-17的產生,還降低IL-10濃度(圖5A)。這些效果與T細胞增殖差異無關。用ACM處理不影響增殖的T細胞的數量(圖5B),不過其影響了預分裂T細胞分裂數量,這可能解釋了降低的IFN-γ和IL-10水平(圖5C)。但是,無論ACM是否含有IL-38,在T細胞增殖方面不存在差異(圖5B,C)。這些數據顯示來自凋亡細胞的IL-38限制了巨噬細胞依賴的TH17細胞的產生并維持IL-10表達。
實施例5:IL-38在凋亡細胞中經N末端加工
除了IL-IRa,IL-1的全部成員都作為前體生成,它們需要在N末端剪切以達到完全的活性。最近,根據N末端前結構域的大小,將IL-38歸類進IL-36亞族中(4,19)。與該亞族的其它成員相同,IL-38具有一致基序,推測這可能決定了N末端切割位點(圖6A)。為了確定凋亡是否誘導IL-38加工,在腫瘤細胞中過表達C末端帶有myc標記的IL-38,并從這些細胞產生ACM。在免疫沉淀IL38后,進行2D凝膠電泳以顯現IL-38亞型。在凝膠中成功地鑒定了兩種IL-38亞型,表明IL-38確實在凋亡期間被加工(圖6B)。挑取兩個代表推測的IL-38亞型的斑點,通過質譜儀(MS)進行分析。在具有較高分子量的斑點中(推測為全長IL-38),在MS分析中發現了兩種N末端肽,一種來自氨基酸9至18,另一種來自氨基酸24至41,而在具有較低分子量的樣品中僅發現來自氨基酸24至41的肽(圖6C)。因此,IL-38在凋亡細胞中是經N末端加工的。
實施例6:全長和截短的IL-38通過IL-1RAPL1對細胞因子產生施加相反的作用
為了確定全長和截短的IL-38是否具有不同的生物學活性,在經單獨IL-1β或者聯合不同濃度的全長(IL-38aal-152)或截短(IL-38aa20-152)的IL-38刺激的人巨噬細胞上清液中檢測IL-6濃度。在IL-β刺激后,較高濃度的IL-38aal-152(20ng/ml,l0ng/ml)顯著增加了IL-6的產生,而IL-38aa20-152甚至以較低濃度施用時也降低IL-6產生(圖7A)。由于ACM中的IL-38通過與IL-lRAPLl相互作用來調節IL-6產生,所以發明人想了解兩種IL-38亞型(isoform)(在細胞因子產生中具有相反作用)是否均結合IL-lRAPLl。發明人進行了受體結合測定,如上文所闡釋的。在本測定中,兩種IL-38亞型都結合IL-lRAPLl(圖7B)。但是,結合動力學似乎稍有區別。當考慮到盡管IL-38aal-152和IL-38aa20-152對細胞因子產生施加相反作用,但它們都能夠結合IL-1RAPL1時,另一待分析的關鍵點為對產生IL-6的影響是否都是IL-lRAPLl依賴的。為此,在巨噬細胞中進行了瞬時IL-lRAPLl敲除,并在單獨IL-1β或聯合IL-38aal-152或IL-38aa20-152刺激后測量了上清液中的IL-6濃度。巨噬細胞中IL-lRAPLl敲除消除了全長IL-38的IL-6誘導和截短的IL-38的IL-6抑制(圖7C)。
實施例7:IL-38調節IL-lRAPLl激活的途徑JNK/AP1
發明人得到了IL-38調節與凋亡細胞相互作用后的巨噬細胞中的AP-1的證據(圖2D)。為了分析IL-38所影響的與其與IL-1RAPL1相互作用有關的信號轉導途徑,發明人首先使用了HEK 293T細胞的受體過表達模型。用IL-1RAPL1的過表達構建體和AP1或NFκB報告基因構建體共轉染該細胞。將經報告基因構建體轉染而沒有IL-1RAPL1過表達的HEK細胞用作對照。首先,為了表征該模型,用IL-1β刺激IL-1RAPL1過表達細胞和對照細胞,并測量AP1(圖8A)或NFκB(圖8B)活性。將IL-1β用作孤兒受體IL-1RAPL1的低親和配體(14)。IL-1β刺激后,在對照細胞中觀察到NFκB而不是AP1活性的顯著誘導。然而,當IL-1RAPL1過表達時,觀察到AP1激活以及增強的NFκB活性。因此,單獨IL-1β以IL-lRAPLl獨立的方式誘導NFκB激活,而不是AP1激活,其需要IL-1RAPL1。有趣的是,甚至沒有任何刺激物,與對照細胞相比,IL-1RAPL1的存在就足以增加AP1和NFκB活性(圖8A,B)。因此,IL-1β刺激后,在HEK細胞中IL-1RAPL1激活AP1而不是NFκB,但是在過表達時誘導AP1和NFκB激活,無需添加外源配體。為了確認這一點,使用了IL-6啟動子構建體,在不同的轉錄因子結合位點(AP1、NFκB、CREB和CEBPβ)中存在或不存在點突變。用IL1RAPL1過表達質粒和IL-6報告基因構建體共轉染HEK細胞(29)。在這種設計中,與HEK對照細胞比較,IL-1RAPL1的過表達還激活IL-6啟動子。當AP1和NFκB結合位點突變時,消除了這種IL-6啟動子誘導(圖8C)。這表明存在由HEK細胞產生的IL-1RAPL1的內源配體。接著,發明人想了解IL-38aal-152和IL-38aa20-152是否能夠影響IL-1RAPL1下游的AP1和NFκB活性。這種設計中,NFκB啟動子活性不受IL-38調節(圖8D),但是AP1誘導由兩種IL-38亞型負調節(圖3E)。重要的是,與全長蛋白比較,IL-38aa20-152能夠在更低的濃度調節AP1激活。為了分析導致IL-38依賴的對IL-lRAPL誘導的AP1活性的抑制的信號轉導機制,與對照HEK細胞相比較在IL-1RAPL1過表達細胞中進行了磷酸化的JNK和p38的細胞內染色。在IL-1RAPL1過表達后,在磷酸化的JNK中觀察到誘導而不是在p38中。這種誘導被IL-38aa20-152顯著降低,而不是IL-38aal-152,證實了截短的IL-38的更強調節作用。
實施例8:IL-38調節巨噬細胞中的AP1活性
接下來,發明人將發明人的數據從HEK模型轉移進巨噬細胞情況中。用AP1或NFκB報告基因構建體轉染人巨噬細胞,并用單獨IL-1β或聯合IL-38aa20-152或IL-38aal-152刺激。如同在HEK細胞中,IL-38aa20-152在巨噬細胞中降低AP1而不是NFκB活性,而IL-38aal-152是無效的(圖9)。因此,只有截短的IL-38抑制巨噬細胞中的AP-1活性,這與發明人發現的經缺乏IL-38的凋亡細胞上清液刺激的巨噬細胞顯示出較高水平的AP1活性一致(圖2D)。最后,發明人著手這個問題:為什么IL-38aal-152在IL-1β刺激巨噬細胞后增加IL-6產生(圖7A),而在HEK細胞模型中,IL-38aal-152既不增強AP1也不增強NFκB激活。為了研究這種差異,用IL-6報告基因構建體轉染巨噬細胞,并用單獨IL-1β或者聯合兩種IL-38亞型來刺激。如所預期的,IL-38aa20-152減弱了IL-6啟動子活性,而IL-38aal-152完全不影響IL-6啟動子誘導(圖9),表明IL-38aal-152介導的IL-6的產生的增加不是在轉錄上調節的。
綜上所述,本發明顯示了經N末端加工的IL-38,其可用在臨床上來限制一般的自身炎癥或者例如有缺陷的巨噬細胞和凋亡細胞相互作用導致的自身炎癥。
參考文獻:
1.Sims,J.E.&Smith,D.E.(2010)Nat Rev Immunol 10,89-102.
2.Dinarello,C.A.(2009)Annu Rev Immunol 27,519-50.
3.Dinarello,C.A.(2013)Seminars in immunology.
4.Garlanda,C,Dinarello,C.A.&Mantovani,A.(2013)Immunity 39,1003-18.
5.Towne,J.E.,Renshaw,B.R.,Douangpanya,J.,Lipsky,B.P.,Shen,M.,Gabel,C.A.&Sims,J.E.(2011)J Biol Chem 286,42594-602.
6.Latz,E.,Xiao,T.S.&Stutz,A.(2013)Nat Rev Immunol 13,397-411.
7.Boraschi,D.&Tagliabue,A.(2013)Seminars in immunology.
8.Pavlowsky,A.,Gianfelice,A.,Pallotto,M.,Zanchi,A.,Vara,H.,Khelfaoui,M.,Val-negri,P.,Rezai,X.,Bassani,S.,Brambilla,D.,Kumpost,J.,Blahos,J.,Roux,M.J.,Humeau,Y.,Chelly,J.,Passafaro,M.,Giustetto,M.,BiUuart,P.&Sala,C.(2010)Current biology:CB 20,103-15.
9.Gambino,F.,Kneib,M.,Pavlowsky,A.,Skala,H.,Heitz,S.,Vitale,N.,Poulain,B.,Khelfaoui,M.,Chelly,J.,BiUuart,P.&Humeau,Y.(2009)The European journal of neuroscience 30,1476-86.
10.Jin,H.,Gardner,R.J.,Viswesvaraiah,R.,Muntoni,F.&Roberts,R.G.(2000)European journal of human genetics:EJHG 8,87-94.
11.Born,T.L.,Smith,D.E.,Garka,K.E.,Renshaw,B.R.,Bertles,J.S.&Sims,J.E.(2000)The Journal of biological chemistry 275,29946-54.
12.Carrie,A.,Jun,L.,Bienvenu,T.,Vinet,M.C,McDonell,N.,Couvert,P.,Zemni,R.,Cardona,A.,Van Buggenhout,G.,Frints,S.,Hamel,B.,Moraine,C,Ropers,H.H.,Strom,T.,Howell,G.R.,Whittaker,A.,Ross,M.T.,Kahn,A.,Fryns,J.P.,Beldjord,C,Marynen,P.&Chelly,J.(1999)Nature genetics 23,25-31.
13.Khan,J.A.,Brint,E.K.,O'Neill,L.A.&Tong,L.(2004)The Journal of biological chemistry 279,31664-70.
14.Pavlowsky,A.,Zanchi,A.,Pallotto,M.,Giustetto,M.,Chelly,J.,Sala,C.&BiUuart,P.(2010)Commun Integr Biol 3,245-7.
15.Arthur,J.S.&Ley,S.C.(2013)Nat Rev Immunol 13,679-92.
16.Ley,S.,Weigert,A.,Heriche,J.K.,Mille-Baker,B.,Janssen,R.A.&Brune,B.(2013)Immunobio logy 218,40-51.
17.Lin,H.,Ho,A.S.,Haley-Vicente,D.,Zhang,J.,Bernal-Fussell,J.,Pace,A.M.,Hansen,D.,Schweighofer,K.,Mize,N.K.&Ford,J.E.(2001)The Journal of biological chemistry 276,20597-602.
18.Bensen,J.T.,Dawson,P.A.,Mychaleckyj,J.C.&Bowden,D.W.(2001)Journal of interferon&cytokine research:the official journal of the International Society for Interferon and Cytokine Research 21,899-904.
19.van de Veerdonk,F.L.,Stoeckman,A.K.,Wu,G.,Boeckermann,A.N.,Azam,T.,Netea,M.G.,Joosten,L.A.,van der Meer,J.W.,Hao,R.,Kalabokis,V.&Dinarello,C.A.(2012)Proc Natl Acad Sci U S A 109,3001-5.
20.Jung,M.Y.,Kang,S.W.,Kim,S.K.,Kim,H.J.,Yun,D.H.,Yim,S.V.,Hong,S.J.&Chung,J.H.(2010)Scandinavian journal of rheumatology 39,190-6.
21.Guo,Z.S.,Li,C,Lin,Z.M.,Huang,J.X.,Wei,Q.J.,Wang,X.W.,Xie,Y.Y.,Liao,Z.T.,Chao,S.Y.&Gu,J.R.(2010)International journal of immunogenetics 37,33-7.
22.Chou,C.T.,Timms,A.E.,Wei,J.C,Tsai,W.C,Wordsworth,B.P.&Brown,M.A.(2006)Annals of the rheumatic diseases 65,1106-9.
23.Monnet,D.,Kadi,A.,Izac,B.,Lebrun,N.,Letourneur,F.,Zinovieva,E.,Said-Nahal,R.,Chiocchia,G.&Breban,M.(2012)Annals of the rheumatic diseases 71,885-90.
24.Rahman,P.,Sun,S.,Peddle,L.,Snelgrove,T.,Melay,W.,Greenwood,C.&Glad-man,D.(2006)Arthritis and rheumatism 54,2321-5.
25.Dehghan,A.,,et al.(2011)Circulation 123,731-8.
26.Zitvogel,L.,Kepp,O.&Kroemer,G.(2010)Cell 140,798-804.
27.Ley,S.,Weigert,A.,Weichand,B.,Henke,N.,Mille-Baker,B.,Janssen,R.A.&Brune,B.(2013)Oncogene 32,631-40.
28.Weigert,A.,Tzieply,N.,von Knethen,A.,Johann,A.M.,Schmidt,H.,Geisslinger,G.&Brune,B.(2007)Mol Biol Cell 18,3810-9.
29.Xiao,W.,Hodge,D.R.,Wang,L.,Yang,X.,Zhang,X.&Farrar,W.L.(2004)Prostate 61,354-70.
- 還沒有人留言評論。精彩留言會獲得點贊!