一種雙相多酶偶聯體系高效制備(S)?2?(4?硝基苯基)環氧乙烷的方法與流程
本發明涉及一種雙相多酶偶聯體系高效制備(S)-2-(4-硝基苯基)環氧乙烷的方法,屬于生物工程技術領域。
背景技術:
(S)-2-(4-硝基苯基)環氧乙烷((S)-p-nitrostyrene oxide,(S)-pNSO)分子式為C8H7NO3,與(R)-2-(4-硝基苯基)環氧乙烷互為對映異構體,是一類在有機合成和藥物制備中非常重要的中間體物質,可用于β-受體阻滯劑硝苯洛爾等藥物的合成。
手性環氧化物的制備方法有拆分法和合成法。
拆分法包括生物拆分法和化學拆分法,由于拆分法固有的限制,得到的產物最大理論得率僅為50%。通過合成法可突破這一限制,得到理論產率為100%的產物。
合成法分為化學合成法和生物合成法。其中,化學合成法以金屬催化的不對稱合成為主,但目前大多數還存在成本太高、反應條件比較苛刻、不綠色環保、收率太低、光學純度不夠等諸多問題。生物合成法則具有選擇性高、反應條件溫和、綠色環保的特點,目前逐漸成為研究熱點。通過把微生物的完整全細胞或者其中的酶當作生物催化劑,可實現物質的轉化,有的反應可通過一種酶一步反應就可以完成,有的需要多種酶和一定反應條件或多種酶幾步反應來完成催化反應。
對于(S)-pNSO合成方法,目前主要集中于化學合成法和化學酶促法,也有通過雙酶串聯法制備得到(S)-pNSO,但是也需要經由化學法得到雙酶的底物。
其中,化學合成法是將α-溴代對硝基苯乙酮溶于適量甲醇中,加入硼氫化鈉還原α-溴代對硝基苯乙酮,得到消旋體2-(4-硝基苯基)環氧乙烷,再通過手性柱制備得到(S)-pNSO,化學制備法生成大量副反應,得到產品產率低,手性柱制備成本高,難以應用于實際工業生產中。
化學酶促法雖然得到的產物(S)-pNSO的光學純度大于99%ee,但是步驟復雜,要通過一步化學合成方法和兩步生物合成法得到終產物(S)-pNSO。首先將α-溴代對硝基苯乙酮溶于甲醇中,加入硼氫化鈉反應一定時間后向反應液中加入水,用乙酸乙酯提取混合物,加入鹵水處理以除去雜質,經干燥、旋蒸去除溶劑,得到消旋體2-溴-1-(4-硝基苯基)乙醇和消旋體2-(4-硝基苯基)環氧乙烷的混合物。將混合物溶解后加入緩沖液,添加含有鹵代醇脫鹵酶的全細胞開始脫鹵閉環反應,待消旋體2-溴-1-(4-硝基苯基)乙醇完全轉化為2-(4-硝基苯基)環氧乙烷,迅速調整pH和溫度,添加亞硝酸鈉及鹵代醇脫鹵酶,得到(S)-pNSO的ee值大于99%,最后通過硅膠色譜柱將產物純化。化學酶促法步驟冗長、后處理過程復雜使得大規模經濟生產(S)-pNSO較為困難。
技術實現要素:
本發明的目的是提供一種雙相多酶偶聯的方法,提高生物合成法制備(S)-pNSO的得率和e.e.值,有較大的工業化生產和應用潛力以及經濟價值。
本發明的技術方案:
(1)將共表達葡萄糖脫氫酶和羰基還原酶的單質粒雙啟動子的菌株E.coli/pETDuet-Sygdh-Sys1發酵后,離心收集菌體,添加到緩沖溶液/異丁醇兩相反應溶液中,同時添加添加一定量的NADP+、葡萄糖、對硝基溴代苯乙酮(BNP),實現不對稱還原對硝基溴代苯乙酮(BNP)產生中間產物(S)-2-溴-1-(4-硝基苯基)乙醇((S)-BNE)的過程;
(2)將單表達鹵代醇脫鹵酶基因的菌株E.coli/pET-28a(+)-SyHheC發酵培養后,破碎菌體,離心收集上清液得到鹵代醇脫鹵酶粗酶液,待步驟(1)中底物BNP轉化為(S)-BNE后,向其中添加鹵代醇脫鹵酶粗酶液,并補充適量緩沖溶液,反應一定時間;
(3)酶促反應結束后,向反應體系中加入異丁醇進行萃取,離心得到萃取相,并干燥、過濾得到(S)-pNSO。
在本發明的一種實施方式中,步驟(1)所述緩沖溶液/異丁醇兩相反應溶液是按體積比7:3或6:4或5:5或4:6或3:7混合的100mmol/L的pH 8.0的磷酸鉀緩沖液與異丁醇。
在本發明的一種實施方式中,步驟(1)所述緩沖溶液/異丁醇兩相反應溶液是按體積比7:3混合的100mmol/L的pH 8.0的磷酸鉀緩沖液與異丁醇。
在本發明的一種實施方式中,步驟(1)底物BPN的初始反應濃度為10-30mmol/L,葡萄糖初始反應濃度為80mmol/L、輔因子NADP+的初始濃度為0.2mmol/L,重組大腸桿菌E.coli/pETDuet-Sygdh-Sys1的用量以濕細胞計為70mg/ml。
在本發明的一種實施方式中,步驟(1)反應溫度為35℃,攪拌轉速為220rpm,反應時間為0.5~9h。
在本發明的一種實施方式中,步驟(1)反應溫度為35℃,攪拌轉速為220rpm,反應時間為4~9h。
在本發明的一種實施方式中,步驟(1)反應時間為9h。
在本發明的一種實施方式中,步驟(2)是向步驟(1)的體系中添加50μl的3.03U/ml的鹵代醇脫鹵酶粗酶液和一定量的磷酸鉀緩沖液,反應180min。
本發明先利用葡萄糖脫氫酶和羰基還原酶催化BNP得到中間產物(S)-BNE,再通過鹵代醇脫鹵酶催化(S)-BNE反應得到(S)-pNSO;本發明通過有機-水兩相反應體系可解決單相溶液系統中底物溶解度差和環氧底物會發生自發水解的問題,利用多酶偶聯的方法,不用經過化學法合成酶的底物,僅需相對便宜的前手性底物、廉價的輔助底物和添加少量的輔酶就可以得到高e.e.值、高收率的目標產品,產物(S)-2-(4-硝基苯基)環氧乙烷摩爾產率可達99.9%,e.e.值>99%。可見,本發明能使生產成本降低,且條件溫和、方法簡便。
附圖說明
圖1:水/有機兩相體系中多酶偶聯催化合成(S)-pNSO的工藝路線;
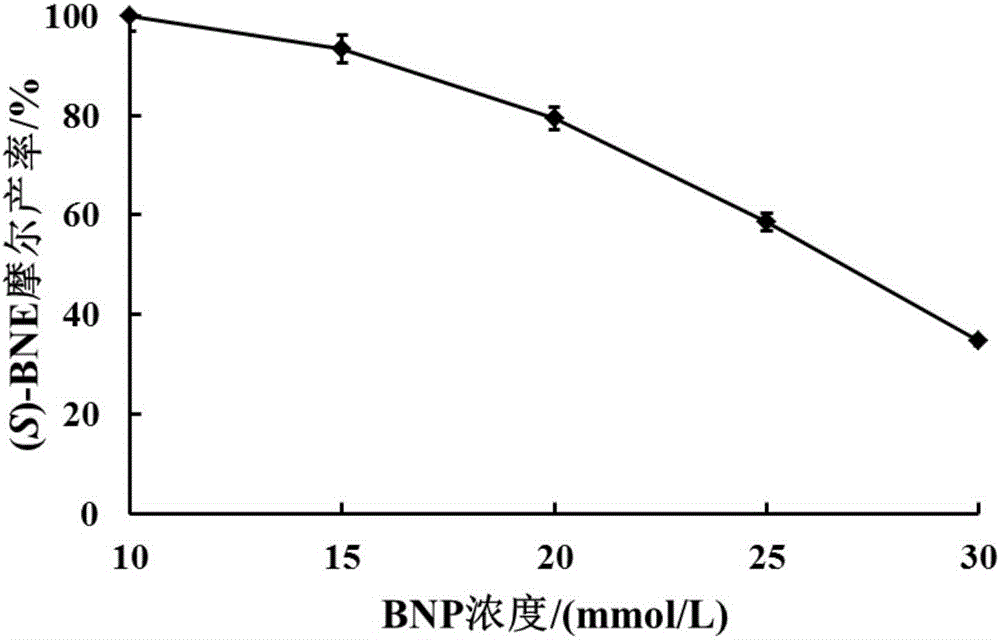
圖2:BNP濃度對多酶催化反應的影響;
圖3:不同配比的緩沖液/異丁醇對多酶催化反應的影響;
圖4:反應時間對合成(S)-pNSO第一步反應的影響;
圖5:反應時間對合成(S)-pNSO第二步反應的影響。
具體實施方式
以2-溴-1-(4-硝基苯基)-乙醇為底物,每分鐘消耗1μmol底物所需的酶量定義為1個鹵代醇脫鹵酶酶活性單位(U)。
產物的檢測方法:在反應體系中加入等體積的異丁醇進行萃取,離心吸取異丁醇相,適量無水硫酸鎂干燥,0.22μm有機膜過濾后進行HPLC檢測。所用HPLC檢測(S)-BNE、(S)-pNSO產率和e.e.值的方法:高效液相色譜(HPLC)為Waters高效液相色譜儀,反相色譜柱為AS-H(4.6mmΦ×250mmL×5μm),柱溫為30℃,采用2489紫外檢測器,檢測波長220nm,一步等度洗脫,流動相為異丙醇/正己烷(8/2,v/v),流速為0.8mL/min,進樣量為10μL。(S)-BNE的出峰時間為15.783min,(S)-pNSO的出峰時間為16.940min,底物BPN出峰時間為22.518min。
計算產物的產率和e.e.值采用外標法計算:分別配置0.25mmol/L、0.5mmol/L、1mmol/L、1.5mmol/L和2mmol/L的(S)-BNE、(S)-pNSO的標準品溶液,以濃度為橫坐標,峰面積為縱坐標作標準曲線。c待=A待×c標/A標,其中c待和A待代表待測樣品的摩爾濃度和峰面積;c標和A標代表標準品的摩爾濃度和峰面積。e.e.值=[(S-R)/(S+R)]×100%:其中S和R分別代表(S)-和(R)-BNE或(S)-和(R)-pNSO的最終摩爾濃度。
實施例1重組菌E.coli BL21/pETDuet-Sygdh-Sys1及E.coli/pET-28a(+)-SyHheC的發酵及菌液、酶液的獲取
共表達葡萄糖脫氫酶和羰基還原酶的單質粒雙啟動子的菌株E.coli/pETDuet-Sygdh-Sys1的構建方法,參見公布號為CN104388373A的、發明名稱為“一種共表達羰基還原酶和葡萄糖脫氫酶的大腸桿菌系統的構建”的中國專利申請。
單表達鹵代醇脫鹵酶基因的菌株E.coli/pET-28a(+)-SyHheC的構建方法參見《食品與生物技術學報》2015年34(5):494-500發表的文章《新型鹵代醇脫鹵酶基因的克隆,序列分析及表達》。是登錄號為KF853591.1的編碼鹵代醇脫鹵酶的基因SyHheC,與表達載體pET28a連接,構建重組表達質粒pET28a-SyHheC,酶切位點為NcoⅠ和NotⅠ,轉化入E.coli BL21(DE3)。
將E.coli BL21/pETDuet-Sygdh-Sys1菌株接種于含終濃度為100μg/mL氨芐青霉素的2mL LB培養基中,37℃、215rpm培養過夜;再分別以2mL/dL轉接入30mL LB培養基中,培養至OD600約為0.8時,加入IPTG至終濃度0.6mmol/L,26℃誘導表達12h。離心培養液(8 000rpm,10min)收集得到E.coli BL21/pETDuet-Sygdh-Sys1濕菌體。
將E.coli/pET-28a(+)-SyHheC菌株接種于含終濃度為100μg/mL卡那霉素的2mL LB培養基中,37℃、215rpm培養過夜;再分別以2mL/dL轉接入30mL LB培養基中,培養至OD600約為0.8時,加入IPTG至終濃度0.1mmol/L,30℃誘導表達8h。離心培養液(8 000rpm,10min)收集得到E.coli/pET-28a(+)-SyHheC菌體后,用6mL磷酸鉀緩沖液(20mmol/L,pH 8.0)懸浮。以功率200W、工作3s、間隔8s的條件冰浴超聲波破碎E.coli/pET-28a(+)-SyHheC菌體,破碎時間為15min。超聲破碎后,冷凍離心機離心(14000rpm,15min),上清即是SyHheC粗酶液。4℃保存備用。
實施例2有機溶劑的選擇
分別選用對三種酶活性影響較少的幾種有機溶劑,包括二甲基亞砜、二甲基甲酰胺、正己烷、環己烷、正辛烷、異辛烷、正庚烷和異丁醇,溶解底物BNP,肉眼觀察底物的溶解情況。由于底物BNP的溶解性非常差,不溶于水和烷類等有機溶劑,只溶于DMF等高極性溶劑,而另一方面對于產物的檢測技術有限,需要高效液相色譜的檢測方法,而高效液相色譜采用的手性色譜柱AS-H是極性填料,對于BNP的助溶劑的要求是不能使用DMF等極性溶劑。故要選擇一種對BNP、(S)-BNE和(S)-pNSO都有很好的溶解性的且對色譜柱無傷害或傷害最小的,又對涉及的三種酶的酶活性影響小的有機溶劑。本發明選定異丁醇作為有機溶劑。
實施例3底物BNP濃度對中間產物生成的影響
向按體積比4:6混合的100mmol/L的pH 8.0磷酸鉀緩沖液/異丁醇中添加終濃度為80mmol/L的D-葡萄糖、0.2mmol/L的NADP+和70mg/mL的E.coli/Sygdh-Sys1濕菌體,構成1mL反應體系,添加不同濃度的BNP(10~30mmol/L)進行酶促反應,35℃、220rpm搖床中反應12h后,加入1mL異丁醇進行萃取,10000rpm,2min離心吸取異丁醇相,適量無水硫酸鎂干燥,0.22μm有機膜過濾。用高效液相色譜對中間產物(S)-BNE的產率和e.e.值進行分析,結果如下圖2所示,當BNP濃度低于15mmol/L時,中間產物(S)-BNE的產率可達90%以上;而隨著底物濃度的繼續加大,產率急速下降。
實施例4緩沖液/異丁醇配比的選擇對中間產物生成的影響
向按體積比4:6混合的100mmol/L的pH 8.0磷酸鉀緩沖液/異丁醇中添加終濃度為15mmol/L的BNP作為底物、80mmol/L的D-葡萄糖、0.2mmol/L的NADP+作為輔酶循環底物和輔酶、70mg/mL的E.coli/Sygdh-Sys1濕菌體,構成1mL反應體系;改變磷酸鉀緩沖液(100mmol/L,pH 8.0)/異丁醇的兩相配比為7:3、6:4、5:5、4:6、3:7,反應體系終體積為1mL,置于35℃、220rpm搖床中反應12h后,加入1mL異丁醇進行萃取,離心吸取異丁醇相,干燥過濾。高效液相色譜對中間產物(S)-BNE的產率和e.e.值進行分析,發現在不同比例的緩沖液/異丁醇反應體系中,如圖3所示,中間產物(S)-BNE的產率隨著有機相異丁醇的增大而下降。酶在有機溶劑中會損失一部分的酶活性,而且菌體需要一定的水相保持流動性而增加底物和酶的接觸,所以選擇緩沖液/異丁醇配比為7:3,此時(S)-BNE的產率最大。
實施例5合成(S)-pNSO第一步的反應時間進程
1mL體系中含體積比為7:3的100mmol/L磷酸鉀緩沖液(pH 8.0)/異丁醇、15mmol/L底物BNP、80mmol/LD-葡萄糖、0.2mmol/LNADP+和E.coli/Sygdh-Sys1濕菌體70mg/mL,于35℃、220rpm分別反應0.5h、1h、2h、3h、4h、5h、6h、7h、8h、9h,加入1mL異丁醇進行萃取,離心吸取異丁醇相,干燥,0.22μm有機膜過濾。高效液相色譜對中間產物(S)-BNE的產率和e.e.值進行分析,如圖4,時間為橫坐標,產率和e.e.值為縱坐標所得曲線即為合成(S)-pNSO第一步的反應時間進程。在反應開始的前6h產物(S)-BNE的摩爾產率增加比較快速;6h后趨于平緩;反應9h后(S)-BNE的摩爾產率可達99.8%;反應過程中e.e.值始終保持在99.9%以上。所以選擇合成(S)-pNSO第一步的最佳時間為9h。
實施例6合成(S)-pNSO第二步的反應時間進程
實施例4的第一步反應完成后,加入50μL的3.03U/ml的SyHheC酶液(每分鐘消耗1μmol中間產物(S)-BNE所需的酶量定義為1個酶活單位U),并添加適量100mmol/L磷酸鉀緩沖液(pH 8.0)至反應終體積為2mL,于35℃、220rpm分別反應30~180min,間隔30min,加入2mL異丁醇進行萃取,離心吸取異丁醇相,適量無水硫酸鎂干燥,0.22μm有機膜過濾。高效液相色譜對中間產物(S)-pNSO的產率和e.e.值進行分析,如圖5,時間為橫坐標,產率和e.e.值為縱坐標所得曲線即為合成(S)-pNSO第二步的反應時間進程。反應前2h反應速率較大,后趨于平穩,3h后(S)-pNSO的摩爾產率可達99.9%,反應過程中e.e.值始終保持在99.9%以上。
雖然本發明已以較佳實施例公開如上,但其并非用以限定本發明,任何熟悉此技術的人,在不脫離本發明的精神和范圍內,都可做各種的改動與修飾,因此本發明的保護范圍應該以權利要求書所界定的為準。
- 還沒有人留言評論。精彩留言會獲得點贊!