一種內源性糖改善米渣蛋白功能特性的方法及其產品與流程
本發明屬于食品科學與工程技術領域,具體涉及一種內源性糖改善米渣蛋白功能特性的方法及其產品。
背景技術:
以大米為原料發酵生產乳酸、淀粉糖、檸檬酸及生化藥品等的糖化工序會產生大量副產物一米渣。米渣中蛋白質的質量分數高達70%(干基),相當于純大米中蛋白質含量的5.8倍。大米蛋白具有良好的氨基酸組成配比,消化率極高,其品質被公認為糧食種子蛋白質中的最佳者,是一種優良的植物蛋白。由于米渣蛋白的低過敏性,非常適用于嬰幼兒食品。此外大米蛋白還具有重要的保健功能。近年來的研究表明,大米蛋白對抗糖尿病、抗膽固醇和抗癌變等都有一定的作用。
由于經歷了長時間的化學和酶解處理,米渣中大米蛋白不再是天然的大米蛋白。溶解度大大降低并失去與之相關的某些功能性質,限制了其在食品中的應用。此外,在實際生產中,不同食品體系對蛋白質功能特性的要求往往不同,如液態乳制品對蛋白乳化性能要求較高;肉制品對蛋白凝膠性能要求較高;而速溶豆奶粉則對分散性和溶解性能要求更高。因此為了擴大米渣的應用范圍,獲取專用性極強的大米蛋白,必須要對米渣蛋白進行改性。
蛋白質的接枝改性就是通過化學或生物技術對蛋白質的結構進行修飾,在側鏈上的活性基團(如-NH2,-OH等)導入一種或多種基團。常用的修飾技術包括對氨基酸殘基的酰化、去酰胺、磷酸化、糖基化(利用美拉德反應)、烷基化、水解以及氧化等。
中國專利201410266430.8《一種米渣蛋白的糖基化改性方法》提供了一種米渣蛋白的糖基化改性方法,但該方法依賴于外源性糖的添加。
技術實現要素:
為解決現有技術的不足,提高米渣蛋白的乳化性、溶解性、熱穩定性、pH以及鹽離子穩定性等功能特性,提高應用范圍,獲取專用性極強的米渣蛋白,并同時提高米渣的綜合利用率,本發明提供了一種內源性糖改善米渣蛋白功能特性的方法及其產品。
本發明所提供的技術方案如下:
一種內源性糖改善米渣蛋白功能特性的方法,包括以下步驟:將米渣酶解得到米渣內源性糖份和米渣蛋白份,再將所述米渣內源性糖份與所述米渣蛋白份進行美拉德反應,得到內源性糖的糖基化米渣蛋白。
進一步的,內源性糖改善米渣蛋白功能特性的方法包括以下步驟:將米渣酶解,得到米渣內源性糖份和米渣蛋白份的混合份,再將所述混合份進行美拉德反應,得到內源性糖的糖基化米渣蛋白。
具體的,酶解過程為:利用高溫α-淀粉酶將米渣中的淀粉水解至糊精和低聚糖。
具體的,酶解之后:將米渣內源性糖份和米渣蛋白份置于蒸餾水中配置成均勻的分散液,調節pH=7,冷凍干燥,得到待反應粉末,所述待反應粉末再進行美拉德反應。
具體的,酶解之后:利用纖維素酶去除混合份中的纖維素類多糖再進行美拉德反應。
優選的,內源性糖改善米渣蛋白功能特性的方法包括以下步驟:
1)米渣糊化后利用高溫α-淀粉酶將米渣中的淀粉水解至糊精和低聚糖,得到米渣內源性糖份和米渣蛋白份的混合份;
2)將步驟1)得到的混合份利用纖維素酶去除纖維素類多糖,得到待分散混合物;
3)將步驟2)得到的待分散混合物置于蒸餾水得到分散液,攪拌均勻,調pH至7.0,冷凍干燥,得到待反應粉末;
4)將步驟3)得到的待反應粉末置于恒溫恒濕培養箱中進行反應,得到內源性糖的糖基化米渣蛋白。
優選的,步驟1)中:糊化時,米渣與水的固液比為1:8~10,米渣的糊化溫度為85~90℃,糊化時間為10~12min;酶解時,高溫α-淀粉酶的添加量為300~400u/mL,酶解時間為1.5~3h,酶解pH值為5.5~6.0,酶解溫度為85~95℃。
優選的,步驟2)中,纖維素酶的添加量為110~120u/g,酶解時間為2.5~3h,酶解pH值為4.0~4.5。
優選的,恒溫恒濕培養箱的溫度為55~65℃、相對濕度為78~80%、培養時間12~24h。
本發明還提供了根據上述內源性糖改善米渣蛋白功能特性的方法制備得到的內源性糖的糖基化米渣蛋白。
本發明所提供的糖基化蛋白質不僅在乳化性能方面得到改善,在溶解性、熱穩定性、pH以及鹽離子穩定性等功能特性方面都有不同程度的提高,對外界環境的變化具有較大的抵抗能力。現有技術中,一般將蛋白質與糖按照一定的比例溶解于選定pH值的緩沖溶液中,分散均勻后進行冷凍干燥,在50-60℃和相對濕度(用飽和KBr維持在79%的相對濕度)下進行的非酶糖基化反應,通過降低溫度來抑制反應的進行。本發明利用美拉德反應對蛋白質進行糖基化改性,提供了是一種簡單快速的方法。本發明所提供的內源性糖改善米渣蛋白功能特性的方法直接將米渣中的淀粉酶解至糖類,得到米渣蛋白和糖類的混合物,進而通過美拉德反應進行糖基化,得到糖基化米渣蛋白,提高米渣的綜合利用率,而不用添加外源糖類。該方法工藝簡單、無環境污染、適合工業化生產的優勢。得到的渣蛋白可作為液態乳制品、肉制品或速溶制品等的原料或添加劑,專用性極強。
附圖說明
圖1是酶處理法米渣蛋白美拉德反應產物的乳化性與乳化穩定性。
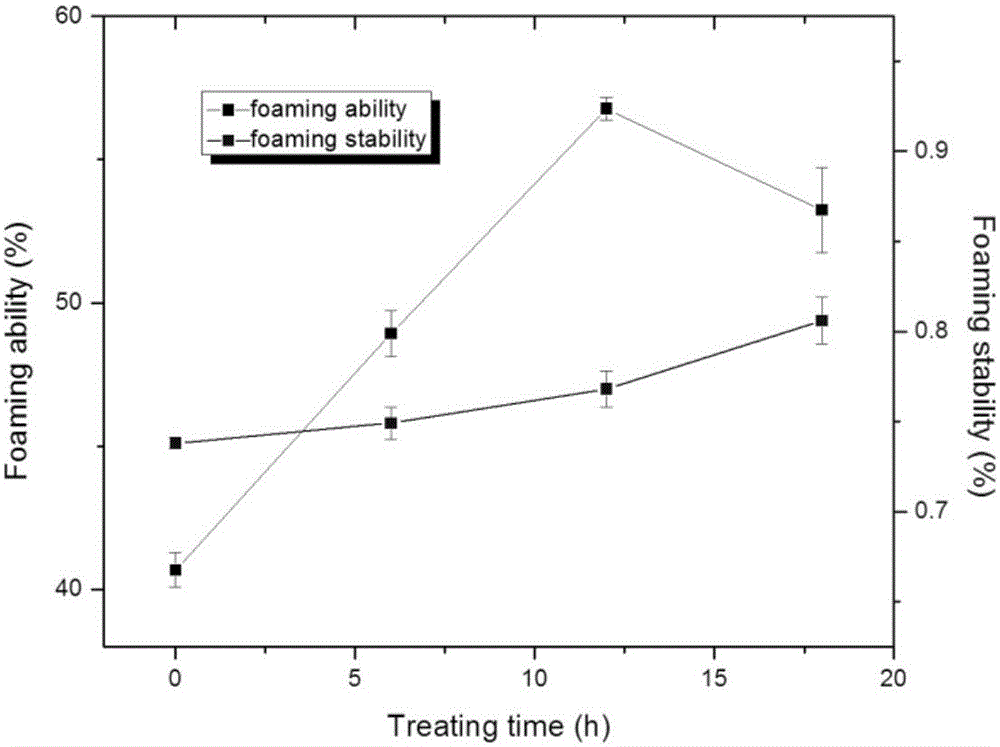
圖2是酶處理法米渣蛋白美拉德反應產物的起泡性和起泡穩定性。
圖3是酶處理法米渣蛋白美拉德反應產物的分散液溶解性對照圖。
圖4為米渣蛋白經過淀粉酶處理后美拉德反應產物在不同pH值條件下電位變化圖。
圖5為米渣蛋白美拉德反應產物電泳圖譜。
圖6為不同pH下米渣蛋白、酶法米渣蛋白改性產物及米渣蛋白-糖復合物的溶解性圖。
圖7為米渣蛋白、酶法米渣蛋白改性產物及米渣蛋白糖復合物的紅外譜圖。
圖8為放大倍數為2000倍的米渣蛋白和內源性糖類-米渣蛋白美拉德法改性產物的掃描電鏡圖。
圖9為放大倍數為1000倍的米渣蛋白和內源性糖類-米渣蛋白美拉德法改性產物的掃描電鏡圖。
圖10為放大倍數為500倍的米渣蛋白和內源性糖類-米渣蛋白美拉德法改性產物的掃描電鏡圖。
具體實施方式
以下對本發明的原理和特征進行描述,所舉實施例只用于解釋本發明,并非用于限定本發明的范圍。
實施例1
將米渣研磨過20目篩,稱取米渣固體粉末10g加入80mL蒸餾水,調節pH至6.0,90℃水浴10min,120r/min搖勻,加入2400u高溫α-淀粉酶,90℃水浴1.5h。冷卻調節pH至4.5加入纖維素酶1100u,55℃水浴2.5h。滅酶,干燥得到混合粉末。將混合粉末分散于蒸餾水,調節體系pH至7.0,冷凍干燥。將冷凍干燥后的粉末平鋪在玻璃或塑料培養皿上,迅速置于恒溫恒濕培養箱中,調節培養箱溫度為60℃,相對濕度為79%,反應12h。本實施例改性米渣蛋白比未改性米渣蛋白相比,乳化性提高了66%,在等電點附近的溶解性提高50%,pH,鹽離子以及熱穩定性明顯改善。
實施例2
將米渣研磨過20目篩,稱取米渣固體粉末50g加入500mL蒸餾水,調節pH至6.0,90℃水浴10min,120r/min搖勻,加入15000u高溫α-淀粉酶,90℃水浴1.5h。冷卻調節pH至4.5加入纖維素酶5500u,55℃水浴2.5h。滅酶,干燥得到混合粉末。將混合粉末分散于蒸餾水,調節體系pH至7.0,冷凍干燥。將冷凍干燥后的粉末平鋪在玻璃或塑料培養皿上,迅速置于恒溫恒濕培養箱中,調節培養箱溫度為60℃,相對濕度為79%,反應12h。本實施例改性米渣蛋白比未改性米渣蛋白相比,乳化性提高了58%,在等電點附近的溶解性提高42%,pH,鹽離子以及熱穩定性明顯改善。
實施例3
將米渣研磨過20目篩,稱取米渣固體粉末10g加入80mL蒸餾水,調節pH至5.5,85℃水浴10min,120r/min搖勻,加入2400u高溫α-淀粉酶,90℃水浴1.5h。冷卻調節pH至4.0加入纖維素酶1100u,55℃水浴2.5h。滅酶,干燥得到混合粉末。將混合粉末分散于蒸餾水,調節體系pH至7.0,冷凍干燥。將冷凍干燥后的粉末平鋪在玻璃或塑料培養皿上,迅速置于恒溫恒濕培養箱中,調節培養箱溫度為55℃,相對濕度為79%,反應12h。本實施例改性米渣蛋白比未改性米渣蛋白相比,乳化性提高了63%,在等電點附近的溶解性提高50%,pH,鹽離子以及熱穩定性明顯改善。
實施例4
將米渣研磨過20目篩,稱取米渣固體粉末10g加入80mL蒸餾水,調節pH至6.0,90℃水浴10min,120r/min搖勻,加入2400u高溫α-淀粉酶,90℃水浴2.0h。冷卻調節pH至4.5加入纖維素酶1100u,55℃水浴3.0h。滅酶,干燥得到混合粉末。將混合粉末分散于蒸餾水,調節體系pH至7.0,冷凍干燥。將冷凍干燥后的粉末平鋪在玻璃或塑料培養皿上,迅速置于恒溫恒濕培養箱中,調節培養箱溫度為60℃,相對濕度為79%,反應15h。本實施例改性米渣蛋白比未改性米渣蛋白相比,乳化性提高了67%,在等電點附近的溶解性提高50%,pH,鹽離子以及熱穩定性明顯改善。
效果例
材料與試劑
米渣(蛋白質70.0%;碳水化合物20.0%;水分9.7%;脂肪3.5%;)堿提米渣蛋白
儀器與設備
試驗方法
乳化性的測定
稱取一定量的蛋白產品溶于50mL蒸餾水中,調節pH值到一定值,加入50mL大豆色拉油,在高速組織搗碎機中均質(10000~12000r/min)2min,再離心(1500r/min)5min。按下式計算乳化能力:
乳化穩定性的測定
將上述離心管置于80℃水浴中,加熱30min后,冷卻至室溫,再離心(1500r/min)5min,測出此時的乳化層高度,則乳化穩定性為:
起泡性和泡沫穩定性的測定
將一定量的蛋白產品溶解到100mL蒸餾水中,調節pH值到一定值,在DS-1高速組織搗碎機中均質(10000~12000r/min)2min,記下均質停止時泡沫體積,則起泡性為:
均質停止30mim后,記下此時泡沫體積,則泡沫穩定性為:
美拉德法改性米渣蛋白的乳化性和乳化穩定性
將采用淀粉酶提取出來的米渣蛋白經過美拉德反應后,分別測定了美拉德反應產物乳化性和乳化穩定性,如圖1所示,0min是指高速分散處理后立即測量的值,10min是指高速分散后放置10min測量的值。從圖1的上半部分可以看出,隨著美拉德反應時間的增加,其乳化性呈現出不斷上升的趨勢;圖1的下半部分表明,隨著美拉德反應時間的增加,其乳化穩定性呈現先降低后升高的趨勢,且在反應12小時以后其乳化穩定性迅速提高。這說明采用美拉德法對米渣蛋白質改性后其乳化性和乳化穩定性得到了改善。
由于在進行美拉德反應時,沒有添加外源性的羰基物質,而是利用在淀粉酶法提取米渣蛋白時水解出的糖類作為糖源。因此合理地利用了資源,而且操作更為簡便。
美拉德法改性米渣蛋白的起泡性和起泡穩定性
將采用淀粉酶提取出來的米渣蛋白經過美拉德反應后,分別測定了美拉德反應產物起泡性和起泡穩定性,如圖2所示。從圖2可以看出,隨著美拉德反應時間的增加,其起泡穩定性呈現出不斷上升的趨勢;隨著美拉德反應時間的增加,其起泡則性呈現先升高后降低的趨勢,在反應12小時呈現最大之后數據急劇下降。因此,在美拉德反應12小時后米渣蛋白質的起泡性性和起泡穩定性得到了改善。
美拉德法改性米渣蛋白分散液的穩定性
如圖3所示,米渣蛋白的等電點(pI)在5.0左右,此時蛋白由于靜電荷為零,靜電斥力減弱導致蛋白溶解性最小,從而使蛋白發生聚集,分散系變渾濁,而美拉德反應之后的米渣蛋白-乳糖共聚物分散系在pH 5.0時比未改性的米渣蛋白澄清(圖3中B)。這可能因為米渣蛋白在美拉德反應后:(1)糖分子的引入,導致靜電作用力增強,足以維持體系的穩定;(2)蛋白質表面多糖的位阻效應對蛋白的聚集有阻礙作用。產物在pH值3-7內保持熱穩定,并且其產物在150mmol/L NaCl離子范圍內保持澄清。說明美拉德反應之后的米渣蛋白-乳糖共聚物溶液能抗高離子強度及酸堿度的改變。圖3中:米渣經過淀粉酶處理后,進行美拉德反應A:0h;B:6h;C:12h;D:18h產物(反應條件:pH 7.0,溫度60℃、濕度79%)(蛋白濃度為5mg/mL)分散液溶解性結果樣品分別調pH至3.0,5.0,7.0;每張圖片從左至右的NaCl濃度為0、10、20、30mmol/L。
美拉德法改性米渣蛋白在不同pH值條件下電位變化如圖4所示,從上到下的曲線依次對應0h、6h、12h、18h。
為了研究清楚到底是蛋白質的表面引入糖基的靜電作用還是其位阻作用導致美拉德反應產物的溶解性及熱穩定性增強,我們對其表面電勢進行了研究,結果顯示:在pH值小于等電點時,米渣蛋白的電位要高于糖基化后產物的電位,在pH值大于等電點時,米渣蛋白的電位高于改性后米渣蛋白-乳糖的電位,其結果表明糖分子接入米渣蛋白后,賴氨酸含量減少,導致正電荷減少,相對負電荷增加。從而說明溶液熱穩定性的增加不是因為靜電作用力增強來維持體系的穩定,而是蛋白質表面多糖的位阻效應對蛋白的聚集有阻礙作用。同時,米渣蛋白在美拉德反應改性后,等電點有所減小。這是因為當還原糖分子的還原末端接入米渣蛋白的游離氨基位點時,減少了米渣蛋白所帶的正電荷,相對增加了米渣蛋白所帶的負電荷,從而導致其等電點向酸性方向偏移。
美拉德法改性米渣蛋白質的電泳特性
圖5為米渣蛋白美拉德反應產物電泳圖譜;
a:蛋白標準品
1:米渣蛋白空白;
2:淀粉酶處理后0h產物;
3:淀粉酶處理后6h產物;
4:淀粉酶處理后12h產物;
5:淀粉酶處理后18h產物;
6:堿法提取米渣蛋白后,蛋白與葡萄糖比1:1;進行美拉德反應6h產物;
7:堿法提取米渣蛋白后,蛋白與葡萄糖比1:1;進行美拉德反應12h產物;
8:堿法提取米渣蛋白后蛋白與葡萄糖比1:1;進行美拉德反應18h產物;(反應條件:pH7.溫度60℃、濕度79%)。
電泳圖譜是分析蛋白質組成成分的重要手段,常使用考馬斯亮藍染色來分析糖基化產物中糖鏈接入后蛋白質分子量的變化。電泳結果如圖5所示,條帶1代表蛋白標準品特征條帶,分子量分布范圍為14.4~116ku。
主要原料與試劑
花生油山東魯花集團有限公司
十二烷基磺酸鈉(SDS)美國Sigma公司
溴化鉀,凱氏定氮試劑,磷酸氫二鈉,磷酸二氫鈉等均為分析純。
主要儀器與設備
pH計FE20 梅特勒—托利多儀器(上海)有限公司
85-2數顯恒溫磁力攪拌器江蘇省金壇市榮華儀器制造有限公司
k9840自動凱式定氮儀濟南海能儀器股份有限公司
離心機長沙平凡儀器儀表有限公司
XHF-D高速分散器寧波新芝科技股份有限公司
UV-5500PC紫外可見分光光度計上海元析儀器有限公司
DSC-Q10型差示掃描量熱儀美國TA公司
S-3000N掃描電子顯微鏡日本HITACHI公司
NEXUS 670傅立葉紅外光譜儀美國尼高力儀器公司
蛋白水解液氨基酸分析系統日本日立公司
J-810圓二色光譜儀日本Jasco公司
溶解性測定
稱取一定量的待測樣品溶于去離子水中,濃度為1mg/mL,用1MHCl或1M NaOH調節溶液的pH值(2、4、6、8、10、12),在磁力攪拌器上常溫攪拌10min,然后4000r/min高速離心10min,取上清液,采用凱式定氮法測定蛋白質含量。
測定米渣蛋白、外源性糖類-米渣蛋白美拉德法改性產物、內源性糖類-米渣蛋白美拉德法改性產物在pH=2、4、6、8、10、12下的溶解性,結果見圖6。
從圖6可看出,對應pH下,與米渣蛋白相比,外源性糖類-米渣蛋白美拉德法改性產物、內源性糖類-米渣蛋白美拉德法改性產物的溶解性都有較大提升,特別是從pH 6開始,提升非常明顯。三者的溶解性均在pH4時最低,此時比較接近米渣蛋白的等電點。外源性糖類-米渣蛋白美拉德法改性產物、內源性糖類-米渣蛋白美拉德法改性產物在較寬的pH范圍內都具有較好的溶解性。外源性糖類-米渣蛋白美拉德法改性產物從pH=8開始變化的趨勢更加明顯,說明它能夠保持較好溶解性的pH范圍比米渣蛋白麥芽糊精復合物更窄,而內源性糖類-米渣蛋白美拉德法改性產物的溶解性表現的更為穩定。
對米渣蛋白、內源性糖類-米渣蛋白美拉德法改性產物及外源性糖類-米渣蛋白美拉德法改性產物進行傅里葉紅外光譜分析,結果見圖7。
當蛋白質和糖分子以共價鍵結合后,在紅外圖譜上的典型特征是在3700~3200cm-1范圍內和1260~1000cm-1范圍內吸收增強,這分別是羥基和碳氧鍵的伸縮振動引起的。
從圖7可看出,3700~3200cm-1范圍內的吸收強度,內源性糖類-米渣蛋白美拉德法改性產物>外源性糖類-米渣蛋白美拉德法改性產物>米渣蛋白,說明米渣蛋白通過與糖類發生美拉德反應增加了羥基數量,且米渣蛋白與內源糖類反應增加的羥基數量比與外源糖類反應得多。
在1260~1000cm-1范圍內的吸收強度,內源性糖類-米渣蛋白美拉德法改性產物>外源性糖類-米渣蛋白美拉德法改性產物>米渣蛋白,說明米渣蛋白通過與糖類發生美拉德反應引入了更多碳氧鍵,且米渣蛋白與麥芽糊精反應引入的碳氧鍵比米渣蛋白與葡萄糖反應的多。
酰胺I帶(-NH彎曲振動,1700~1600cm-1)和酰胺III帶(C-N伸縮振動和N-H彎曲振動,1300~1450cm-1)的吸收強度,內源性糖類-米渣蛋白美拉德法改性產物>外源性糖類-米渣蛋白美拉德法改性產物>米渣蛋白,說明米渣蛋白通過與葡萄糖、麥芽糊精發生美拉德反應,其中的-NH2基團發生了反應。
從以上結論綜合可以得出:內源性糖類對米渣蛋白進行美拉德法改性時,羰氨反應程度更高。
掃描電子顯微鏡(SEM)分析
對米渣蛋白和內源性糖類-米渣蛋白美拉德法改性產物進行掃描電子顯微鏡分析,結果見圖8、圖9、圖10。
在圖8、圖9、圖10中,每張圖中左右部分依次是米渣蛋白和內源性糖類-米渣蛋白美拉德法改性產物。
從放大倍數2000的掃描電鏡圖中可看出:米渣蛋白呈螺旋結構有序地纏繞在一起,結構較稀松;米渣蛋白與內源性糖類發生美拉德反應后米渣蛋白充分伸展開來。
從放大倍數1000的掃描電鏡圖中可看出:米渣蛋白結構有序且稀松;米渣蛋白與內源性糖類發生美拉德反應后米渣蛋白呈片狀形態。
從放大倍數500的掃描電鏡圖中可看出:米渣蛋白以圓條狀有序纏繞在一起;米渣蛋白與內源性糖類發生美拉德反應后部分米渣蛋白充分伸展開來,變成片狀形態,與沒有充分伸展開來的米渣蛋白包圍在一起。
綜上所述可以得出結論,米渣蛋白與內源性糖類發生美拉德反應后部分米渣蛋白的二級結構遭到破壞,分子充分伸展開來,喪失了原有形態,呈片狀形態。
綜合以上各部分結論,結果表明:采用內源性糖類對米渣蛋白進行改性時,美拉德反應進行的更為徹底。
以上所述僅為本發明的較佳實施例,并不用以限制本發明,凡在本發明的精神和原則之內,所作的任何修改、等同替換、改進等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!