一種改性的聚縮酮PCADK及制備方法和醫用用途與流程
技術領域
本發明提供一種改性的聚縮酮PCADK,同時還公開了制備方法和醫用用途,屬于藥用材料領域。
背景技術:
蛋白質藥物是一類新型的用于腫瘤治療與預防的生物藥物,與傳統的化學藥物相比,蛋白類藥物作為治療藥物具有藥效顯著、靶點專一、副作用低、品種繁多等優點,有望成為未來臨床治療的重要發展方向。但是,由于蛋白質為生化制劑,其半衰期短、清除率高、滲透力差、易受環境中的復雜理化因素影響而失去其生物活性,使其作為治療藥物表現出較差的穩定性和治療效果。因此,高效的運輸載體是蛋白類藥物應用的關鍵。
目前,生物可降解材料主要包括淀粉、明膠、葡聚糖、聚乳酸(PLA)和聚乳酸-乙醇酸共聚物(PLGA)等,其中,PLGA最為常用。PLGA緩釋周期長,避免頻繁給藥,且使用安全,生物利用度顯著提高,可應用于注射、口服、埋植等劑型。但PLGA體內降解產物為二氧化碳和水,酸性物質的產生會影響蛋白類藥物的穩定性,帶來后期不釋放等問題。
聚縮酮家族,目前包括PPADK,PCADK及PK1-PK3。作為一類新型生物可降解材料,具有良好的生物兼容性、pH敏感性和蛋白友好性。與廣泛應用的聚酯類高分子不同,聚縮酮在堿性或中性環境中相對穩定,在酸性條件下快速降解為無毒、中性小分子代謝產物,不改變環境pH值從而引起炎癥反應,有效的避免了聚酯類降解產物產生的酸性環境對蛋白類藥物穩定性的影響,解決了蛋白變性后釋放困難等問題,彌補了聚酯類材料的不足。同時,能夠對體內不同部位pH值的變化做出回應,是蛋白類藥物靶向腫瘤(pH 6.0-7.0)及溶酶體(pH 4.0-6.0)等部位的理想載體。
但由于聚縮酮分子量偏低,力學性能較差,單獨形成藥物載體時外表易形成塌陷、結構松散、多孔洞、導致藥物包封效率極低,只能采取與PLGA混用的辦法,來提高載體的剛性結構和包封效率。此外,腫瘤靶向制劑通常以靜脈注射方式給藥,聚縮酮作為疏水性高聚物,進入人體血液系統后,將不可避免的吸附血漿蛋白,進而被巨噬細胞識別吞噬,因此,聚縮酮的生物兼容性有待提高。
聚乙二醇修飾pH敏感性聚合物,可以增加材料的親水性,增加腫瘤細胞對載體的攝取,同時提高聚縮酮的分子量,改善聚縮酮的力學性能,使其能夠自包封單獨形成藥物載體,增加蛋白類藥物的包封效率。此外,聚乙二醇化技術仍有諸多優勢,如降低免疫原性、延長載體循環時間及增加穩定性等。
技術實現要素:
本發明提供一種改性的聚縮酮PCADK及制備方法,解決了現有聚縮酮PCADK力學性質差、分散穩定性差及生物兼容性不好等問題,對聚縮酮PCADK進行親水性修飾,從而獲得一系列蛋白友好型的pH敏感雙親性材料。
一般地,材料表面親水性越好,在血液環境中分散性越好,對血漿蛋白的阻抗能力越強,被免疫系統清除的概率越小,同時,作為雙親性材料,在溶液中自組裝形成載體的能力更強。本發明選用聚乙二醇修飾聚縮酮,通過活性化合物作為中間連接分子,通過共價鍵與聚縮酮末端羥基連接,形成的氨酯鍵在酸性環境下為可降解化學鍵。與此同時,親水性的聚乙二醇阻礙血漿蛋白的特異性吸附,延長載體材料在體內的血液循環周期,提高載體材料的分散穩定性。
本發明的另一目的是提供上述聚縮酮PCADK改性材料的制備方法。
本發明所述聚縮酮PCADK改性材料的結構式為:
其中,n = 31 ~ 34, 是聚乙二醇單甲醚鏈段,分子量分別為2000,5000和10000,m分別對應為45,113和227。a-c聚合物分別對應聚縮酮-聚乙二醇單甲醚2000,聚縮酮-聚乙二醇單甲醚5000和聚縮酮-聚乙二醇單甲醚10000。
本發明所述的一種聚縮酮PCADK改性材料的制備方法,步驟如下:
1)聚縮酮PCADK的合成
7.25mmol 的1, 4-環己烷二甲醇溶解在20 ~ 30mL蒸餾苯中,加熱到90 ~ 100°C。35 ~ 55mg對甲苯磺酸溶解在0.5 ~ 1mL乙酸乙酯中,加入到上述苯溶液;加入7.25mmol的2, 2-二甲氧基丙烷,開始反應;每隔2h補償2mL的苯和500μL 的2, 2-二甲氧基丙烷,氮氣保護下攪拌反應24 ~ 48h,然后加入100 ~ 200μL三乙胺停止反應,合成的聚縮酮在0.5 ~ 1L冷的正己烷中沉淀分離;
2)聚乙二醇單甲醚的活化
聚乙二醇單甲醚的末端羥基的活化,0.2mmol的聚乙二醇單甲醚溶解在5~ 10mL的氯仿中,加入60 ~ 100倍摩爾比的1, 6-亞甲基二異氰酸酯,加熱到60 ~ 80°C,過夜攪拌回流;粗產物在1~2L沉淀劑中多次沉淀,即獲得活化的聚乙二醇單甲醚;
3)目標產物的合成
將步驟 (2) 獲得的活化的0.2mmol聚乙二醇單甲醚溶解在6~8mL的氯仿中,0.8 ~ 1.2倍摩爾量的聚縮酮溶解在2 ~ 4mL氯仿中,聚縮酮溶液逐滴加入上述溶液,加熱到60 ~ 80°C,過夜攪拌回流;粗產物在1 ~ 2L沉淀劑中多次沉淀,即獲得目標產物。
本發明所述聚縮酮PCADK改性材料的制備方法,其特征在于:
所述步驟2)中聚乙二醇單甲醚分別是聚乙二醇單甲醚2000,聚乙二醇單甲醚5000和聚乙二醇單甲醚10000時,對應的產物分別為聚縮酮PCADK-聚乙二醇單甲醚2000,聚縮酮PCADK-聚乙二醇單甲醚5000和聚縮酮PCADK-聚乙二醇單甲醚10000。
本發明所述聚縮酮PCADK改性材料的制備方法,其特征在于:
所述步驟2)、3) 沉淀劑選自正己烷、石油醚、甲基叔丁基謎、乙醚、1,4-二氧六環等有機溶劑。
本發明的積極效果在于:
提供一種改性的聚縮酮PCADK,解決了現有聚縮酮PCADK力學性質差、分散穩定性差及生物相容性不好等問題,對聚縮酮進行親水性修飾,從而獲得一系列蛋白友好型的pH敏感雙親性材料,具有良好的pH敏感性、親水性、穩定性和力學性能,能夠獨立作為藥物載體使用,包封化藥及蛋白類藥物,靶向腫瘤及炎癥部位。
附圖說明
圖1為實施例1聚縮酮PCADK改性材料的合成示意圖;
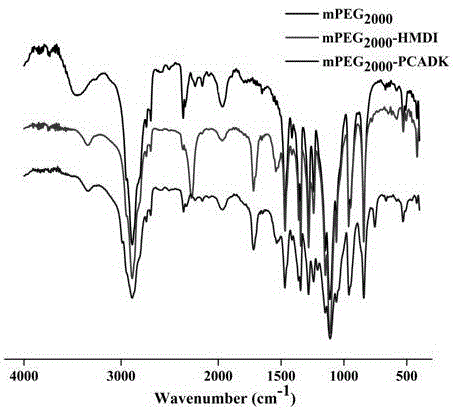
圖2為實施例1聚縮酮PCADK改性材料的傅里葉紅外光譜圖;
圖3為實施例1聚縮酮PCADK改性材料的凝膠滲透色譜圖;
圖4為實施例1 ~ 3聚縮酮PCADK改性材料的細胞毒性結果;
圖5為實施例1 ~ 3聚縮酮PCADK改性材料制備人血清白蛋白(HSA)納米粒的穩定性結果。
具體實施方式
通過以下實施例進一步舉例描述本發明,并不以任何方式限制本發明,在不背離本發明的技術解決方案的前提下,對本發明所作的本領域普通技術人員容易實現的任何改動或改變都將落入本發明的權利要求范圍之內。
實施例1
聚縮酮PCADK-聚乙二醇單甲醚2000,其結構式為:
其制備方法如下:
(1)聚縮酮PCADK的合成
1, 4-環己烷二甲醇溶解在20 ~ 30mL蒸餾苯中,加熱到90 ~ 100°C。35 ~ 55mg對甲苯磺酸溶解在0.5 ~ 1mL乙酸乙酯中,加入到上述苯溶液。加入與1, 4-環己烷二甲醇等摩爾量的2, 2-二甲氧基丙烷,開始反應。隨機補償蒸餾的苯和2, 2-二甲氧基丙烷,氮氣保護下攪拌反應24 ~ 48h,然后加入100 ~ 200μL三乙胺停止反應,合成的聚縮酮在0.5 ~ 1L冷的正己烷中沉淀分離。
(2)聚乙二醇單甲醚2000的活化
聚乙二醇單甲醚2000的末端羥基的活化,聚乙二醇單甲醚2000溶解在5 ~ 10mL氯仿中,加入60 ~ 100摩爾倍數的1, 6-亞甲基二異氰酸酯,加熱到60 ~ 80°C,過夜攪拌回流。粗產物在1 ~ 2L正己烷中多次沉淀,即獲得活化的聚乙二醇單甲醚2000。
(3)目標產物的合成
將步驟 (2) 獲得的活化的聚乙二醇單甲醚2000溶解在5 ~ 10mL氯仿中,等摩爾量的聚縮酮 PCADK溶解在少量氯仿中,聚縮酮溶液逐滴加入上述溶液,加熱到60 ~ 80°C,過夜攪拌回流。粗產物在1 ~ 2L正己烷中多次沉淀,即獲得聚縮酮PCADK-聚乙二醇單甲醚2000。
聚縮酮PCADK-聚乙二醇單甲醚2000傅立葉紅外譜圖見圖2,凝膠滲透色譜圖見圖3。如圖2所示,中間產物紅外譜圖同時存在1, 6-亞甲基二異氰酸酯特征峰及氨酯鍵特征峰,說明聚乙二醇單甲醚2000與1, 6-亞甲基二異氰酸酯的成功連接,終產物紅外譜圖1, 6-亞甲基二異氰酸酯特征峰的消失與氨酯鍵特征峰的存在,說明聚乙二醇單甲醚2000與聚縮酮PCADK的成功連接。
實施例2
聚縮酮PCADK-聚乙二醇單甲醚5000,其結構式為:
其制備方法如下:
(1)聚縮酮PCADK的合成
1, 4-環己烷二甲醇溶解在20 ~ 30mL蒸餾苯中,加熱到90 ~ 100°C。35 ~ 55mg對甲苯磺酸溶解在0.5 ~ 1mL乙酸乙酯中,加入到上述苯溶液。加入與1, 4-環己烷二甲醇等摩爾量的2, 2-二甲氧基丙烷,開始反應。隨機補償蒸餾的苯和2, 2-二甲氧基丙烷,氮氣保護下攪拌反應24 ~ 48h,然后加入100 ~ 200μL三乙胺停止反應,合成的聚縮酮在0.5 ~ 1L冷的正己烷中沉淀分離。
(2)聚乙二醇單甲醚5000的活化
聚乙二醇單甲醚5000的末端羥基的活化,聚乙二醇單甲醚5000溶解在5 ~ 10mL氯仿中,加入60 ~ 100摩爾倍數的1, 6-亞甲基二異氰酸酯,加熱到60 ~ 80°C,過夜攪拌回流。粗產物在1 ~ 2L正己烷中多次沉淀,即獲得活化的聚乙二醇單甲醚5000。
(3)目標產物的合成
將步驟 (2) 獲得的活化的聚乙二醇單甲醚5000溶解在5 ~ 10mL氯仿中,等摩爾量的聚縮酮PCADK溶解在少量氯仿中,聚縮酮溶液逐滴加入上述溶液,加熱到60 ~ 80°C,過夜攪拌回流。粗產物在1 ~ 2L正己烷中多次沉淀,即獲得聚縮酮PCADK-聚乙二醇單甲醚5000。
實施例3
聚縮酮PCADK-聚乙二醇單甲醚10000,其結構式為:
其制備方法如下:
(1)聚縮酮PCADK的合成
1, 4-環己烷二甲醇溶解在20 ~ 30mL蒸餾苯中,加熱到90 ~ 100°C。35 ~ 55mg對甲苯磺酸溶解在0.5 ~ 1mL乙酸乙酯中,加入到上述苯溶液。加入與1, 4-環己烷二甲醇等摩爾量的2, 2-二甲氧基丙烷,開始反應。隨機補償蒸餾的苯和2, 2-二甲氧基丙烷,氮氣保護下攪拌反應24 ~ 48h,然后加入100 ~ 200μL三乙胺停止反應,合成的聚縮酮在0.5 ~ 1L冷的正己烷中沉淀分離。
(2)聚乙二醇單甲醚10000的活化
聚乙二醇單甲醚10000的末端羥基的活化,聚乙二醇單甲醚10000溶解在5 ~ 10mL氯仿中,加入60 ~ 100摩爾倍數的1, 6-亞甲基二異氰酸酯,加熱到60 ~ 80°C,過夜攪拌回流。粗產物在1 ~ 2L乙醚/正己烷中多次沉淀,即獲得活化的聚乙二醇單甲醚10000。
(3)目標產物的合成
將步驟 (2) 獲得的活化的聚乙二醇單甲醚10000溶解在5 ~ 10mL氯仿中,等摩爾量的聚縮酮PCADK溶解在少量氯仿中,聚縮酮溶液逐滴加入上述溶液,加熱到60 ~ 80°C,過夜攪拌回流。粗產物在1 ~ 2L正己烷中多次沉淀,即獲得聚縮酮PCADK-聚乙二醇單甲醚10000。
實驗例1
細胞毒性實驗:
人類乳腺癌細胞株MCF7培養于含10% 胎牛血清、1%青霉素-鏈霉素的DMEM細胞培養液中,培養瓶置于37°C,含5% CO2培養箱中。取對數生長期的細胞進行以下實驗。
以1×104個/孔的密度將處于對數期的MCF7細胞接種于在96孔板中,37°C培養24h使其貼壁,第二天去除培養液,分別加入100mg/mL預先制備的含聚縮酮改性材料的培養基,繼續培養24h。去除培養基,每孔加入20μL、5mg/mL MTT試劑,37°C孵育4h,DMSO溶解生成的甲瓚結晶,酶標儀492nm分析溶液吸光度,并計算細胞存活率。
實驗結果如圖4所示,聚縮酮-聚乙二醇單甲醚(PCADK-mPEG)對MCF7細胞24h后的殺傷作用明顯低于聚縮酮PCADK。這歸功于表面聚乙二醇單甲醚的修飾,降低其材料毒性,屏蔽聚縮酮表面負電荷,增加材料的親水性和生物兼容性。
實驗例2
聚縮酮改性材料制備HSA納米粒:
采用雙乳化溶劑揮發法制備HSA納米粒。200μL的20mg/mL的HSA逐滴加入含有40mg聚縮酮或聚縮酮改性材料的2mL二氯甲烷中,高壓均質剪切機22000rpm均質1.5min形成初乳,初乳立即注入到20mL含有1%PVA的外水相,高壓均質剪切機22000rpm均質2min,形成的復乳過夜磁力攪拌揮發有機溶劑。去離子水離心洗滌三次,獲得的HSA納米粒-40°C條件下冷凍干燥,4°C保存。
實驗例3
HSA納米粒穩定性表征:
稱取冷凍干燥后的聚縮酮或聚縮酮改性材料制備HSA納米粒20mg,重懸在2mL pH 7.4 PBS中,靜置三周,觀察沉淀或聚集現象。
實驗結果如圖5所示,圖A為靜置一天現象,圖B為靜置三周現象,聚縮酮 PCADK組EP管底部明顯有沉淀現象,而聚縮酮修飾材料組依舊呈懸浮狀態,說明聚縮酮修飾材料親水性和穩定性較好。
實驗例4
HSA納米粒的粒徑及電位分布檢測:
采用激光粒度儀分析HSA納米粒粒徑及電位分布,HSA納米粒測試濃度0.5mg/mL。實驗結果如表1所示,HSA納米粒平均粒徑基本在200 ~ 300 nm之間,電位在 -2 ~ 0 mV之間。親水性的修飾提高了聚縮酮表面電位,同時也增加了在溶液中的粒徑,這歸功于聚乙二醇單甲醚的親水作用。
表1.實施例1 ~ 3聚縮酮PCADK改性材料制備納米粒粒徑及電位分布
實驗例5
HSA納米粒的包封效率檢測:
采用BCA蛋白濃度試劑盒檢測納米粒的包封效率。20mg的納米粒溶解于1mL,1M的NaOH溶液放置一天,隨后加入2mL,1M的HCl溶液放置一天,最后NaOH溶液中和降解液。降解液中的蛋白濃度稀釋后使用BCA蛋白試劑盒分析。實驗結果如表1所示,納米粒的實際包封效率分布在 73.2%~ 85.8%之間。聚縮酮PCADK改性材料包封率稍低于聚縮酮PCADK,可能歸功于聚縮酮改性材料的親水性,在納米粒的制備過程中導致蛋白藥物趨于分布于納米粒的表面,從而降低了藥物包封效率。
- 還沒有人留言評論。精彩留言會獲得點贊!