一種鹽酸溴己新的制備方法與流程
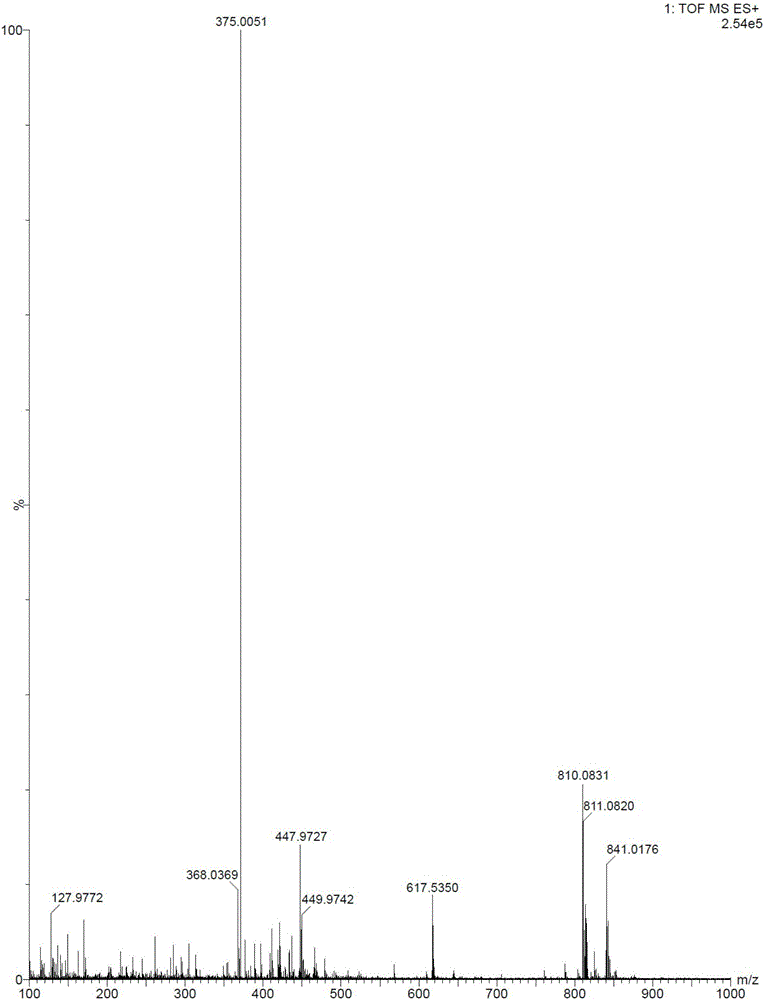
本發明屬于藥物合成領域,具體涉及一種鹽酸溴己新的制備方法。
背景技術:
:鹽酸溴己新(BromhexineHydrochloride,N-(2-氨基-3,5-二溴芐基)-N-甲基環己胺鹽酸鹽)是由勃林格殷格翰公司研制的藥物,臨床上用于急慢性支氣管炎、哮喘、支氣管擴張、肺氣腫的治療,尤其適用于白色粘痰咳出的困難者,及因痰液廣泛阻塞小支氣管引起的氣急等癥。目前,關于鹽酸溴己新的合成工藝文獻報道比較多,而綜合文獻分析發現,報道的鹽酸溴己新的合成方法包括以下幾種,其合成路線如下:路線一路線二路線三路線一的工藝中涉及的縮合劑大多為二環己基碳二亞胺(DCC),其副產物二環己基脲(DCU)毒性大,去除難度較大,其殘留風險影響鹽酸溴己新的用藥安全。路線二的反應類型均較為簡單,但工藝步驟長,操作繁瑣,生產周期長;而且工藝過程中涉及安全風險較大的鹵代試劑,對反應設備的要求較高;同時,鹵代試劑活性較高,可能導致苯環上的溴原子被取代而產生副反應,影響產品質量。雖然路線三的工藝較為簡便:醋酸催化條件下,加熱、甲苯分水進行縮合反應,縮短了生產周期。但是該路線的甲苯分水步驟需要高溫反應,度反應設備要求高;同時還存在反應不完全的風險,工業化生產中存在安全風險。綜上,提供一種鹽酸溴己新的制備方法,工藝簡單,操作簡便,反應條件溫和,反應收率較好,有較好的工業化基礎成為了本領域技術人員亟待解決的問題。技術實現要素:本發明提供了一種鹽酸溴己新的制備方法,該制備方法工藝簡單,操作簡便,反應條件溫和,收率好,純度高,雜質含量少,所需原料易得,具有良好的工業化基礎。本發明所述的一種鹽酸溴己新的制備方法,包括如下步驟:a:2-氨基-3,5-二溴苯甲醛、N-甲基環己胺與鈦酸酯在溶劑中反應生成過渡態;b:還原劑原位還原所述過渡態,生成溴己新游離堿;c:所述溴己新游離堿成鹽制備鹽酸溴己新;反應式為:優選地,所述R為C1-C6的直鏈烷基或支鏈烷基。進一步優選地,所述直鏈烷基為甲基、乙基、正丙基、正丁基,所述支鏈烷基為異丙基、異丁基。優選地,所述步驟a中,所述2-氨基-3,5-二溴苯甲醛與所述N-甲基環己胺的摩爾比為1:0.8~1:4,所述2-氨基-3,5-二溴苯甲醛與所述鈦酸酯的摩爾比為1:1~1:3。進一步優選地,所述2-氨基-3,5-二溴苯甲醛與所述N-甲基環己胺的摩爾比為1:2~1:3。優選地,所述步驟a中,所述溶劑選自醇類溶劑、醚類溶劑、鹵代烴類溶劑、腈類溶劑和亞砜類溶劑中的一種或幾種。進一步優選地,所述溶劑選自為甲醇、乙醇、異丙醇、正丁醇、四氫呋喃、1,4-二氧六環、二氯甲烷中的一種或多種。優選地,所述步驟b中,所述還原劑選自硼氫化鈉、硼氫化鉀、硼氫化鋰中的一種或幾種,所述2-氨基-3,5-二溴苯甲醛與所述還原劑的摩爾比為1:0.3~1:3,所述步驟a、所述步驟b、所述步驟c的反應溫度為-10~50℃。進一步優選地,所述步驟a、所述步驟b、所述步驟c的反應溫度為20~30℃。與現有技術相比,本發明的有益效果:本發明以2-氨基-3,5-二溴苯甲醛和N-甲基環己胺為原料,經還原胺化、成鹽制備鹽酸溴己新。采用本發明制備鹽酸溴己新,縮短了鹽酸溴己新的反應步驟,且所需原料易得。本發明中,2-氨基-3,5-二溴苯甲醛結構中的醛基在-10~50℃的溫度下能與N-甲基環己胺和鈦酸酯發生反應產生含有縮醛結構的過渡態,所得過渡態無需分離純化,可直接被還原劑還原生成溴己新游離堿。該工藝可在同一反應容器中進行,避免了現有技術中需對中間產物進行分離或純化的步驟。本發明工藝簡單,操作簡便,反應條件溫和,所需原料易得,有利于工業化生產。采用本發明制備鹽酸溴己新收率好,純度高,雜質含量少,能夠保證藥品的安全、有效。附圖說明圖1為鹽酸溴己新的高分辨質譜圖。圖2為鹽酸溴己新的核磁共振氫譜圖。具體實施方式以下具體實施方式可以對本發明的內容做出更為詳細的說明,但本發明的主題范圍不局限于以下具體實施例,凡是基于本
發明內容所實現的技術、工藝均屬于本發明的范圍。實施例1本實施例中的原料均來源于市售。將2.0g2-氨基-3,5-二溴苯甲醛、1.2gN-甲基環己胺溶于20ml甲醇中,室溫攪拌條件下將4g鈦酸四異丙酯緩慢滴入到反應液中,加完后繼續室溫攪拌反應5h;將0.3g硼氫化鈉分批加入反應液中,室溫攪拌反應2h。TLC檢測反應完全。反應結束后,向反應液中加入20ml水淬滅反應,析出大量固體,過濾,濾餅用乙酸乙酯洗滌3次,合并濾液和洗液,乙酸乙酯萃取得有機相,依次用碳酸鈉溶液、飽和食鹽水洗滌有機相。室溫、攪拌條件下,向上述有機相中滴入5ml6N鹽酸,攪拌析晶3h。過濾得一白色固體,乙醇重結晶、干燥,得到2.3g鹽酸溴己新,收率:78%。HRMS-ESI(m/z):375.0051(M+H+);1H-NMR(400MHz,CD3OD)δ=7.70(d,1H),7.48(d,1H),4.50(d,1H),4.23(d,1H),3.40~3.34(m,1H),2.70(s,3H),2.18~2.09(m,2H),1.97(s,2H),1.76~1.57(m,3H),1.47~1.37(m,2H),1.27~1.24(m,1H)ppm.實施例2本實施例中的原料均來源于市售。將2.0g2-氨基-3,5-二溴苯甲醛、4gN-甲基環己胺溶于20ml甲醇中,于10℃攪拌條件下將2g鈦酸四異丙酯緩慢滴入到反應液中,加完后繼續于10℃攪拌反應5h;將0.3g硼氫化鈉分批加入反應液中,室溫攪拌反應2h。TLC檢測反應完全。反應結束后,向反應液中加入20ml水淬滅反應,析出大量固體,過濾,濾餅用乙酸乙酯洗滌3次,合并濾液和洗液,乙酸乙酯萃取得有機相,依次用碳酸鈉溶液、飽和食鹽水洗滌有機相。于10℃攪拌條件下,向上述有機相中滴入10ml6N鹽酸,攪拌析晶3h。過濾得一白色固體,乙醇重結晶、干燥,得到2.1g鹽酸溴己新,收率:71%。實施例3本實施例中的原料均來源于市售。將2.0g2-氨基-3,5-二溴苯甲醛、1.2gN-甲基環己胺溶于20ml異丙醇中,于30℃攪拌條件下將4g鈦酸四異丙酯緩慢滴入到反應液中,加完后繼續室溫攪拌反應5h;將0.3g硼氫化鈉分批加入反應液中,于30℃攪拌反應2h。TLC檢測反應完全。反應結束后,向反應液中加入20ml水淬滅反應,析出大量固體,過濾,濾餅用乙酸乙酯洗滌3次,合并濾液和洗液,乙酸乙酯萃取得有機相,依次用碳酸鈉溶液、飽和食鹽水洗滌有機相。于30℃攪拌條件下,向上述有機相中滴入5ml6N鹽酸,攪拌析晶3h。過濾得一白色固體,乙醇重結晶、干燥,得到2.4g鹽酸溴己新,收率:81%。實施例4本實施例中的原料均來源于市售。將2.0g2-氨基-3,5-二溴苯甲醛、1.2gN-甲基環己胺溶于20ml甲醇中,于0℃攪拌條件下將4.9g鈦酸四丁酯緩慢滴入到反應液中,加完后繼續于0℃攪拌反應6h;將0.3g硼氫化鈉分批加入反應液中,室溫攪拌反應2h。TLC檢測反應完全。反應結束后,向反應液中加入20ml水淬滅反應,析出大量固體,過濾,濾餅用乙酸乙酯洗滌3次,合并濾液和洗液,乙酸乙酯萃取得有機相,依次用碳酸鈉溶液、飽和食鹽水洗滌有機相。于0℃攪拌條件下,向上述有機相中滴入5ml6N鹽酸,攪拌析晶3h。過濾得白色固體,乙醇重結晶、干燥,得到2.0g鹽酸溴己新,收率:68%。實施例5本實施例中的原料均來源于市售。將2.0g2-氨基-3,5-二溴苯甲醛、2gN-甲基環己胺溶于20ml1,4-二氧六環中,于-10℃攪拌條件下將4g鈦酸四異丙酯緩慢滴入到反應液中,加完后繼續于-10℃攪拌反應8h;將0.3g硼氫化鈉分批加入反應液中,室溫攪拌反應4h。TLC檢測反應完全。反應結束后,向反應液中加入20ml水淬滅反應,析出大量固體,過濾,濾餅用乙酸乙酯洗滌3次,合并濾液和洗液,乙酸乙酯萃取得有機相,依次用碳酸鈉溶液、飽和食鹽水洗滌有機相。于-10℃攪拌條件下,向上述有機相中滴入5ml6N鹽酸,攪拌析晶3h。過濾得白色固體,乙醇重結晶、干燥,得到2.3g鹽酸溴己新,收率:79%。實施例6本實施例中的原料均來源于市售。將2.0g2-氨基-3,5-二溴苯甲醛、1.2gN-甲基環己胺溶于20ml甲醇中,于50℃攪拌條件下將4g鈦酸四異丙酯緩慢滴入到反應液中,加完后繼續于50℃攪拌反應5h;將0.5g硼氫化鉀分批加入反應液中,室溫攪拌反應2h。TLC檢測反應完全。反應結束后,向反應液中加入20ml水淬滅反應,析出大量固體,過濾,濾餅用乙酸乙酯洗滌3次,合并濾液和洗液,乙酸乙酯萃取得有機相,依次用碳酸鈉溶液、飽和食鹽水洗滌有機相。于50℃攪拌條件下,向上述有機相中滴入5ml6N鹽酸,攪拌析晶3h。過濾得白色固體,乙醇重結晶、干燥,得到2.2g鹽酸溴己新,收率:74%。實施例7對比例:采用
背景技術:
中路線一方法制備鹽酸溴己新。將11g2-氨基-3,5-二溴苯甲酸、2g4-二甲胺基吡啶和5gN-羥基丁二酰亞胺(HOSu)溶于20mL二氯甲烷中,攪拌條件下將溶有10g二環己基碳二亞胺(DCC)的二氯甲烷溶液緩慢滴入上述反應液中,繼續攪拌反應,析出大量固體,繼續反應2h。將18gN-甲基環己胺加入反應液中,攪拌反應2h。TLC監控反應完全。向反應液中加入10mL6NHCl淬滅反應,過濾,萃取得有機相。攪拌條件下,向上述有機相中分批加入1.2gLiBH4,加完后,加熱至40℃反應。TLC監控反應完全。反應結束后,向反應液中加入20ml水淬滅反應,二氯甲烷萃取得有機相,依次用碳酸鈉溶液、飽和食鹽水洗滌有機相。攪拌條件下,向上述有機相中滴入20ml6N鹽酸,攪拌析晶3h。過濾得白色固體,乙醇重結晶,得到9.5g鹽酸溴己新,收率:62%。實施例8對比例:采用
背景技術:
中路線二的方法制備鹽酸溴己新。11.2g2-氨基-3,5-二溴苯甲醛、20ml無水乙醇,攪拌下分批次加入0.95g、NaBH4,控制溫度在10~40℃,非均相反應,TLC監控反應完全。反應完畢后,向反應體系中加入50ml水,攪拌下滴加2N鹽酸調溶液pH=5~6,繼續攪拌30min,將固體過濾,濾餅用水(20ml×3)洗滌,干燥,得還原產物為白色固體。將上述還原產物分批加入到30ml氯化亞砜中,滴加過程中保持溫度0~40℃,滴加完畢后常溫下攪拌反應,反應時間3~4小時。反應完畢后,將過量的氯化亞砜常溫下減壓濃縮,殘余物中加入20ml石油醚,攪拌20min,過濾,石油醚(10ml×3)洗滌,30℃下減壓干燥,得氯帶中間體為黃色固體。將上述氯帶中間體分批加入到11.34gN-甲基環己胺中,再加入20ml無水乙醇,常溫下攪拌反應。TLC監控反應進程,反應時間3~4小時。反應完畢后,將反應液中的溶劑減壓濃縮,向殘余物中加入40ml乙酸乙酯,有固體析出,繼續攪拌20min,將固體過濾,濾液用HCl/EA溶液跳pH=5~6,降溫至0~5℃下攪拌析晶,過濾,干燥,得到鹽酸溴己新粗品,甲醇重結晶得到鹽酸溴己新約6.8g,收率:41%。實施例9對比例:采用
背景技術:
中路線三的方法制備鹽酸溴己新。依次將110gN-甲基環己胺、90g2-氨基-3,5-二溴苯甲醇加入到反應瓶中,攪拌條件下向反應液中加入50g冰醋酸和60mL甲苯,加熱分水(內溫約110~120℃,外溫約130~140℃),TLC監控反應進度至原料不再減少。減壓濃縮后,加入120mL乙醇溶解,攪拌條件下滴加6NHCl至溶液體系pH為2,冷卻析晶過夜,過濾得到鹽酸溴己新粗品,精制得鹽酸溴己新約93g,收率:70%實施例10鹽酸溴己新中雜質的測定。取實施例1-9所得鹽酸溴己新,測定雜質含量。實驗方法:照《中國藥典》(2015年版,二部)鹽酸溴己新【檢查】項下有關物質測定方法測定,結果見下表。表1雜質含量測定表最大單一雜質(%)總雜質(%)實施例10.0210.033實施例20.0370.058實施例30.0320.069實施例40.0430.062實施例50.0200.044實施例60.0410.072實施例7(對比例)0.1150.215實施例8(對比例)0.0760.144實施例9(對比例)0.0920.185由表1可知,本發明的最大單一雜質和雜質總量都優于工藝路線一、工藝路線二和工藝路線三。綜上,采用本發明所得的鹽酸溴己新的收率更高,雜質含量更少。且本發明工藝簡單,操作簡便,反應條件溫和,所需原料易得,具有良好的工業化基礎。上述實施例僅為本發明的優選實施方式之一,不應當用于限制本發明的保護范圍,但凡在本發明的主體設計思想和精神上作出的毫無實質意義的改動或潤色,其所解決的技術問題仍然與本發明一致的,均應當包含在本發明的保護范圍之內。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1