去糖化的海參次級皂苷及其制備方法與流程
本發明涉及一種海參皂苷的酶解制備方法,具體涉及利用糖苷酶酶解海參皂苷的方法,最終得到脫除兩個糖基的海參次級皂苷。
背景技術:
海參為棘皮動物門、海參綱、楯手目動物。海參皂苷是海參體內重要的次生代謝產物同時也是重要的活性物質,海參皂苷主要活性作用——抗腫瘤、抗真菌、抗病毒、抗衰老以及溶血作用,還有調節特定細胞的生長發育和免疫調節等作用。因此,海參皂苷是海參藥用價值的重要指標。
皂苷具有溶血作用,皂苷可與紅細胞膜結合形成甾醇復合物,從而導致通透性增加,造成血紅蛋白的流失。這一性質增加了皂苷的毒性,阻礙其藥用價值的發揮。Yibing Wang等人研究發現植物皂苷的溶血活性與其分子結構有很大關系——相同苷元,糖鏈上糖基個數越多溶血活性越強;相同糖基個數,但糖基之間連接方式不同溶血活性也不同,如雙糖以(1-2)-,(1-4)-,(1-6)-方式連接的皂苷,其溶血性依次增強;糖基上的乙酰化降低皂苷溶血活性。Han M等人進行體內、體外實驗表明,一些糖基化的大量皂苷,如人參皂苷 Rb1、Rb2、Rc,在小腸內吸收非常微弱,而一些脫糖基化的稀有皂苷,則更容易作為在體內發揮作用的活性成分被吸收到血液系統中。并且一些稀有人參皂苷,例如Rg3、Rh2、Rh1、化合物C-K及人參苷元等較未降解前的人參皂苷具有更好的腫瘤細胞毒活性。
海參皂苷的溶血活性比人參皂苷還要強,主要原因可能是海參皂苷的化學結構比人參皂苷復雜,尤其是C-3位置的糖鏈結構的差異。董平等人通過酸水解海參皂苷并研究其活性時發現天然狀態下存在的皂苷成分,溶解生物膜的功效更強,當糖鏈部分少于2個單糖時,其溶血活性會急劇降低,當糖鏈部分含有1個單糖的皂苷或者無糖鏈的苷元,溶血作用很小,在濃度高達200 mg / L時,仍不能使紅細胞完全溶解。學者Kalinin等的研究結果表明:海參皂苷末端為3-O-MeGlc時可促進K+的流失,而其C-4硫酸酯化后則可降低溶血作用,并降低K+流失的速率。
目前,對皂苷的降解方法主要包括化學降解法和酶及微生物降解法。酸水解是最常用,也是操作最為簡便易行的皂苷降解方法。強酸水解可以將糖鏈部分或全部降解掉,得到苷元,但是反應過于激烈,容易發生側鏈的脫水、環和、雙鍵位移等不理想的產物。溫和酸水解往往可以得到多種糖鏈長度不同的次級苷,部分次級苷相對于皂苷會發生構型的轉變。皂苷類化合物分子結構中的糖苷鍵在堿水溶液中降解較難進行,因此通常需要提高反應溫度等加以實驗。酶水解具有一定的專屬性,不同性質的酶作用于不同構型、不同組成糖的糖苷鍵,從而達到定向水解的目的,是最理想的制備次級皂苷的方法。
目前,有很多關于通過酶解作用將高含量的人參皂苷轉化成高活性的稀有人參皂苷的報道,例如,高娟等人通過糖苷酶酶解Rb1制備次級苷Rg3、Rh2、Rh1、化合物CK等次級皂苷,并且有研究證明這些稀有人參皂苷具有更好的抗腫瘤細胞毒活性等生物活性。
然而,對海參皂苷酶解的相關研究很少,因此高娟論文中酶解方法對研究糖苷酶對海參皂苷的酶解作用有非常好的借鑒和指導意義。常用化學方法水解皂苷,但是具有副反應多,產物雜,專一性差等缺點。因此研究新的酶解海參皂苷方法,可以豐富海參皂苷的結構,尤其是非天然存在或在海參體內含量很少的結構類型,為海參皂苷結構、活性等方面的理論和應用研究奠定基礎。
技術實現要素:
本發明的目的之一是提供去糖化的海參次級皂苷,目的之二是提供該海參次級糖苷的具體酶解制備方法,以彌補現有技術的不足。
為達到上述目的,本發明采取的具體技術方案為:
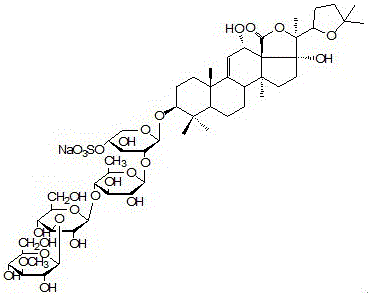
去糖化的海參次級皂苷是海參皂苷Holothurin A和海參皂Echinoside A的糖鏈上由于糖苷酶酶解脫去后兩個糖基后得到的次級皂苷;海參皂苷Holothurin A(如圖1所示),簡稱HA,和海參皂Echinoside A(如圖2所示),簡稱EA。
上述的海參次級皂苷的制備方法包括以下步驟:
⑴、將干海參烘干并粉碎成粉末;
⑵、取上述干海參粉,乙醇浸提后,減壓濃縮得到浸提膏,再凍干得到粗提物;
⑶、將上述粗提物水溶解,再用水飽和正丁醇進行多次萃取;然后取上清液減壓蒸發得到浸提膏,再水浴蒸干成粉末,得到粗皂苷粉;
⑷、利用大孔樹脂柱層析法對上述得到的粗皂苷進行除雜富集,使用乙醇液進行洗脫,檢測洗脫液,將含有皂苷的洗脫液減壓濃縮,并水浴蒸干成粉末,即為總皂苷;
⑸、正相硅膠柱層析純化上述得到的總皂苷,檢測洗脫液,將含有皂苷的洗脫液減壓濃縮,并水浴蒸干,得到富含HA和EA的總皂苷;
⑹、反相硅膠柱層析分離上述富含HA和EA的總皂苷,檢測洗脫液,根據洗脫時間,得到兩種組分,再分別將對應洗脫液減壓濃縮,并水浴蒸干,即得到HA和EA;
⑺、將上述得到的HA和EA分別進行酶解,選用糖苷酶,溫度為40-60℃,pH4-6,添加糖苷酶量根據反應物的量來添加,反應時間為3-7d,將反應液進行水飽和正丁醇萃取,將上層萃取液進行減壓濃縮,水浴蒸干分別得到:去糖化的海參HA次級皂苷和去糖化的海參EA次級皂苷。
進一步的,上述步驟⑶中水飽和正丁醇萃取皂苷5次,第一次使用粗皂苷水溶液:水飽和正丁醇1:3(v/v)萃取,再兩次按1:2(v/v)萃取,最后兩次按1:1(v/v)萃取。
進一步的,上述步驟⑷中乙醇洗脫具體為:大孔樹脂用量與樣品量的比例是30-80(ml):1(g),先用50%(v/v)的乙醇溶液洗脫,后用80%(v/v)乙醇溶液洗脫。
進一步的,上述步驟⑸中正相硅膠柱層析的洗脫條件為:正相硅膠用量與樣品量的比例是30-70(g):1(g),分別用9:1:0.1,8:1:0,7:1:0.1,7:2:0.2(v/v/v)梯度比例的氯仿:甲醇:水進行洗脫,每個梯度洗脫至少5個柱體積,通過進行TLC(展開劑氯仿:甲醇:水,(v/v/v),10%-15%(v/v)硫酸乙醇溶液為顯色劑,在110℃烘箱顯色檢測,每個梯度洗出的雜質在不同位置顯色,再更換為7:2.5:0.2梯度洗脫至TLC能檢測到皂苷,再更換為7:3:0.3梯度洗脫至TLC檢測不再顯色。
進一步的,上述步驟⑹中反相硅膠柱層析的洗脫條件為:反相硅膠用量與樣品量的比例是30-70(g):1(g),先用40%(v/v)的甲醇洗脫,再用60%(v/v)的甲醇溶液洗脫,洗脫液進行減壓濃縮,用TLC(展開劑70%(v/v)甲醇,10%-15%(v/v)硫酸乙醇溶液為顯色劑,在110℃烘箱顯色)法檢測洗脫液,洗脫液減壓濃縮,并60℃水浴蒸干得到HA和EA兩種主要皂苷。
進一步的,上述步驟⑺中,所述糖苷酶為果膠酶或復合纖維素酶。
本發明的有益效果:本發明所使用的糖苷酶在溫和的條件下將海參皂苷酶解成次級皂苷,從而降低海參皂苷的毒性,提高其生物活性;同時,使用糖苷酶酶解海參皂苷,避免了使用微生物降解或從微生物中提取的粗酶進行降解而帶來的副反應及副產物,提高了酶解產物的純度和產量,可以得到單一產物,純度可達100%。另外,本發明提供的制備方法也克服了化學方法反應條件劇烈,苷元部分的脫水、環化、雙鍵轉移等副反應等缺點。本發明具有反應條件溫和,專一性強,產物純度高,副反應少,產物單一,轉化率高等特點。本發明制得去糖化的次級海參皂苷具有低毒性、高活性等的特點,從而促進海參皂苷在醫藥方面的應用。
附圖說明
圖1是海參皂苷Holothurin A分子式化學結構圖。
圖2是海參皂苷Echinoside A分子式化學結構圖。
圖3是實施例1中Pectinex Ultra SP--L酶解HA的薄層層析圖。
圖4是實施例1中Pectinex Ultra SP--L酶解EA的薄層層析圖。
圖5是實施例1中Pectinex Ultra SP--L酶酶解HA的TIC圖。
圖6是實施例1中Pectinex Ultra SP--L酶解HA的質譜圖。
圖7是實施例1中Pectinex Ultra SP--L酶酶解EA的TIC圖。
圖8是實施例1中Pectinex Ultra SP--L酶酶解EA的質譜圖。
圖9是實施例2中Celluclast 1.5 L酶酶解HA的薄層層析圖。
圖10是實施例2中Celluclast 1.5 L酶酶解EA的薄層層析圖。
圖11是實施例2中Celluclast 1.5 L酶酶解HA的TIC圖。
圖12是實施例2中Celluclast 1.5 L酶酶解HA的質譜圖。
圖13是實施例2中Celluclast 1.5 L酶酶解EA的TIC圖。
圖14是實施例2中Celluclast 1.5 L酶酶解EA的質譜圖。
具體實施方式
以下通過具體實施例結合附圖對本發明進一步解釋和說明。
實施例1:
⑴、將購買的干海參放入50℃烘箱,2天后取出,粉碎成粉末;
⑵、取干海參粉(菲律賓刺參約1kg),經過60%乙醇浸提7天,期間不停攪拌,浸提3次,減壓蒸發得到浸提膏,凍干得到粗提物;
⑶、將上述粗提物先用少量水充分溶解,而后用水飽和正丁醇進行五次萃取,第一次按樣品與水飽和正丁醇1:3(v/v)萃取,后兩次按1:2(v/v)萃取,最后兩次按1:1(v/v)萃取。取上清液減壓濃縮得到浸提膏,并用60℃水浴蒸干得到粗皂苷粉;
⑷、用大孔樹脂柱層析法對⑶步中的粗皂苷進行富集,大孔樹脂用量與樣品量的比例是80(ml):1(g),先用50%(v/v)的乙醇溶液洗脫,后用80%(v/v)乙醇溶液洗脫,收集80%乙醇洗脫液,用TLC(展開劑氯仿:甲醇:水,7:3:0.3(v/v/v),10%-15%(v/v)硫酸乙醇溶液為顯色劑,在110℃烘箱顯色)法檢測,將顯示紫紅色斑點(TLC 檢測呈陽性 )的洗脫液減壓濃縮,并60℃水浴蒸干得到總皂苷;
⑸、正相硅膠柱層析純化⑷的總皂苷,正相硅膠用量與樣品量的比例是50(g):1(g),以9:1:0.1- 7:3:0.3(v/v/v)梯度比例的氯仿:甲醇:水依次進行洗脫,用TLC(展開劑氯仿:甲醇:水,7:3:0.3(v/v/v),10%-15%(v/v)硫酸乙醇溶液為顯色劑,在110℃烘箱顯色)檢測,將顯示紫紅色斑點(TLC 檢測呈陽性 )的洗脫液減壓濃縮,并60℃水浴蒸干,得到富含HA和EA的總皂苷;
⑹、ODS(50μm)反相硅膠柱層析分離上述富含HA和EA的總皂苷,ODS (50μm)反相硅膠用量與樣品量的比例是40(g):1(g),先用40%(v/v)的甲醇洗脫,再用60%(v/v)的甲醇溶液洗脫,洗脫液進行減壓濃縮,用TLC(展開劑70%(v/v)甲醇,10%-15%(v/v)硫酸乙醇溶液為顯色劑,在110℃烘箱顯色)法檢測洗脫液,洗脫液減壓濃縮,并60℃水浴蒸干得到HA和EA兩種主要皂苷。
⑺、將上述得到的HA和EA分別進行酶解:
①取上述兩種皂苷單體各5mg;
②添加10μL酶液(Pectinex Ultra SP--L (果膠酶) 購自Novozymes公司,酶活力26000PGU/mL),用醋酸緩沖液(pH5.0)配置成5mL的反應體系;
③在50℃下保溫反應。反應開始時記為0 h,總反應72h,每24h取1mL樣品,用TLC(展開劑氯仿:甲醇:水,7:3:0.3(v/v/v),10%-15%(v/v)硫酸乙醇溶液為顯色劑,在110℃烘箱顯色)檢測;
④將每次取出的反應液,用水飽和正丁醇溶液按1:1(v/v)萃取,取上清液,由于產物量比較少,采用液氮吹干,得到去糖化的海參HA次級皂苷和去糖化的海參EA次級皂苷。
實施例2:
⑴、將購買的干海參放入50℃烘箱,2天后取出,粉碎成粉末;
⑵、取干海參粉(菲律賓刺參約1kg),經過60%乙醇浸提7天,期間不停攪拌,浸提3次,減壓蒸發得到浸提膏,凍干機凍干得到粗提物;
⑶、將上述粗提物先用少量水充分溶解,而后用水飽和正丁醇進行五次萃取,第一次按樣品與水飽和正丁醇1:3(v/v)萃取,后兩次按1:2(v/v)萃取,最后兩次按1:1(v/v)萃取。取上清液減壓濃縮得到浸提膏,并用60℃水浴蒸干得到粗皂苷粉;
⑷、用大孔樹脂柱層析法對⑶步中的粗皂苷進行富集,大孔樹脂用量
與樣品量的比例是80(ml):1(g),先用50%(v/v)的乙醇溶液洗脫,后用80%(v/v)乙醇溶液洗脫,收集80%乙醇洗脫液,用TLC(展開劑氯仿:甲醇:水,7:3:0.3(v/v/v),10%-15%(v/v)硫酸乙醇溶液為顯色劑,在110℃烘箱顯色)法檢測,將顯示紫紅色斑點(TLC 檢測呈陽性 )的洗脫液減壓濃縮,并60℃水浴蒸干得到總皂苷;
⑸、正相硅膠柱層析純化⑷的總皂苷,正相硅膠用量與樣品量的比例是50(g):1(g),以9:1:0.1- 7:3:0.3(v/v/v)梯度比例的氯仿:甲醇:水依次進行洗脫,用TLC(展開劑氯仿:甲醇:水,7:3:0.3(v/v/v),10%-15%(v/v)硫酸乙醇溶液為顯色劑,在110℃烘箱顯色)檢測,將顯示紫紅色斑點(TLC 檢測呈陽性 )的洗脫液減壓濃縮,并60℃水浴蒸干,得到富含HA和EA的總皂苷;
⑹、ODS(50μm)反相硅膠柱層析分離上述富含HA和EA的總皂苷,ODS (50μm)反相硅膠用量與樣品量的比例是40(g):1(g),先用40%(v/v)的甲醇洗脫,再用60%(v/v)的甲醇溶液洗脫,洗脫液進行減壓濃縮,用TLC(展開劑70%(v/v)甲醇,10%-15%(v/v)硫酸乙醇溶液為顯色劑,在110℃烘箱顯色)法檢測洗脫液,洗脫液減壓濃縮,并60℃水浴蒸干得到HA和EA兩種主要皂苷。
⑺、將上述得到的HA和EA分別進行酶解:
①取上述兩種皂苷單體各5mg;
②添加200μL酶液(Celluclast 1.5 L(復合纖維素酶)購自Novozymes公司,酶活力700EGU/g,密度1.2g/mL),用醋酸緩沖液(pH5.0)配置成5mL的反應體系;
③在50℃下保溫反應。反應開始時記為0 h,總反應72h,每24h取1mL樣品,用TLC(展開劑氯仿:甲醇:水,7:3:0.3(v/v/v),10%-15%(v/v)硫酸乙醇溶液為顯色劑,在110℃烘箱顯色)檢測;
④將每次取出的反應液,用水飽和正丁醇溶液按1:1(v/v)萃取,取上清液,由于產物量比較少,采用液氮吹干,得到去糖化的海參HA次級皂苷和去糖化的海參EA次級皂苷。
次級皂苷產物分析:選用HPLC-ESI-MS檢測
用30%乙腈(色譜級)溶液復溶去糖化的海參HA次級皂苷和去糖化的海參EA次級皂苷,得到去糖化的海參HA次級皂苷和去糖化的海參EA次級皂苷溶液。
色譜條件:
流動相:A:乙腈
B;0.1% NH4HCO3
洗脫梯度:0-5-20-30-35-36 (min)
30%-30%-50%-60%-30%-30% (A相)
流速:1mL/min 進樣量:20μL
色譜柱:XDB-C18(4.6×150mm,5μm) 柱溫:30℃
結果分析
①由TLC薄層層析圖可以看出,隨酶解時間的延長,Pectinex Ultra SP--L (果膠酶)對皂苷的酶解程度加深(如圖3、4),到72h時,HA完全轉化為去糖化的次級皂苷,EA部分轉化,可以得出,HA相較于EA酶解轉化效率更高,EA則需要更長時間才能進行全部轉化。
在負離子模式ESI-MS模式下,顯示準分子離子峰m / z 859 [M-Na]-(質譜圖6),所對應的TIC總離子流如圖5所示,可知在13min左右出現離子峰是去糖化的海參HA次級皂苷,并且HA的轉化率達到100%;在負離子模式ESI-MS模式下,顯示準分子離子峰m / z 845 [M-Na]-(質譜圖8),所對應的TIC總離子流如圖7所示,可知在16min左右出現離子峰是EA,在21min左右出現離子峰是去糖化的海參EA次級皂苷,并且EA的轉化率超過了50%。
②由TLC薄層層析圖可以看出,隨著時間的延長,Celluclast 1.5 L(復合纖維素酶)對皂苷的酶解作用加深(如圖9、10),到72h時HA、EA都部分轉化為產物,但是HA較于EA酶解效果要好些。
在負離子模式ESI-MS模式下,顯示準分子離子峰m / z 859 [M-Na]-(質譜圖12),所對應的TIC總離子流如圖11,可知在13min左右出現離子峰是去糖化的海參HA次級皂苷,并且HA的轉化率超過了50%;在負離子模式ESI-MS模式下,顯示準分子離子峰m / z 845 [M-Na]-(質譜圖14),所對應的TIC總離子流如圖13,可知在16min左右出現離子峰是EA,在21min左右出現離子峰是去糖化的海參EA次級皂苷,并且EA的轉化率小于50%。
在酶解過程中,出現了隨著時間的延長,水溶液越來越渾濁,最后生成沉淀的現象。經HPLC-ESI-MS檢測沉淀物為酶解產物,這說明HA和EA酶解產物不易溶于水。
上述實例不作為對本發明的限定,凡是在本發明的范圍內所做的任何修改、等同替換、改進等,均屬于本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!