一種18F標記的炔雌醇及其制備方法和應用與流程
本發明涉及藥物化學領域,具體涉及一種18f標記的炔雌醇及其制備方法和應用。
背景技術:
乳腺癌是女性易患的常見癌癥,易發生轉移,發病率逐年上升且呈年輕化,嚴重影響女性健康。臨床研究發現,雌激素受體陽性乳腺癌的浸潤性行為較低,且對內分泌治療較為敏感。但經過一階段內分泌治療后,病灶的雌激素受體活性可發生改變進而影響后續療效。免疫病理檢查只能獲取手術病灶的雌激素受體分布情況,而原發灶受體的情況并不能完全代表轉移灶,尤其是治療病灶雌激素受體的活性。因此,在早期正確的判斷乳腺癌治療過程中各部分病灶的雌激素受體活性狀況,是及時調整治療方案和建立個體化治療的關鍵。
正電子發射型計算機斷層顯像(positronemissioncomputedtomography,簡稱pet),是核醫學領域比較先進的臨床檢查影像技術。目前,pet在腫瘤、冠心病和腦部疾病這三大類疾病的診療中尤其顯示出重要的價值。16α-18f-17β-雌二醇(16α-[18f]-fluoro-17β-estradiol,18f-fes)是一種雌二醇衍生物,能特異性地與雌激素受體結合,作為雌激素受體的分子影像探針,通過pet/ct顯像能動態、定量和無創傷地反映體內雌激素受體表達水平和分布情況,為乳腺癌患者提供一種有力評價方法。乳腺癌患者的18f-fespet和pet/ct顯像的臨床實驗結果發現,乳腺癌對18f-fes的吸收與雌激素受體的表達水平有明顯的相關性,而且,抗雌激素治療后18f-fes的吸收明顯減少。隨著18f-fespet和pet/ct在女性乳腺癌領域的應用進一步擴展,18f-fes的合成已成為亟需解決的問題。
1984年,daleo.kiesewetter等人報道了在16α-17β-雌二醇的五元環上直接引入[18f]氟離子進行標記實驗,但是該方法標記條件苛刻,需要在-78℃下加入氫化鋁鋰進行反應,這給標記實驗增加了很大的難度和危險性,不具有通用性。1995年,jojannesromer等人改善了標記條件,首先將16α-17β-雌二醇引入磺酰基,然后在磺酰基上引入[18f]氟離子進行標記,雖然反應條件相對溫和,但是該標記反應需要兩步才能完成,時間較長,放射化學產率較低。如何對現有的雌二醇衍生物的合成路線進行改進并標記以最大限度地利用pet技術清楚判斷雌激素受體活性情況,為患者及時調整治療方案,建立個體化治療,已成為本領域亟待解決的一大技術難題。
技術實現要素:
本發明要解決的技術問題在于克服現有技術中18f標記的炔雌醇合成和標記反應條件苛刻且在用藥過程存在嚴重不良反應的缺陷,從而提供一種療效安全、易于合成和標記,并能利用pet技術以清楚判斷雌激素受體活性情況的18f標記的炔雌醇。
為此,本發明提供了一種18f標記的炔雌醇,具有式(i)所示的結構,
其中,r1選自烷基或烷氧基。
優選的,r1為亞乙基或3,6,9,12-四氧雜十四烷基。
本發明提供了一種制備18f標記炔雌醇的方法,包括如下步驟:
(1)取炔雌醇溶解于與水互溶性較好有機溶劑中,將式a-i所示的化合物加入上述的炔雌醇溶液中,然后加入cuso4·5h2o的水溶液、naasc的水溶液和三(2-苯并咪唑基甲基)胺的水溶液,于40-50℃下反應0.5-2h,將所得反應液旋干,加入與水互溶性較好有機溶劑并離心,取上清液,即得式a-ii所示的化合物;
(2)將所述式a-ii所示的化合物溶于n,n-二甲基甲酰胺中,然后加入18f-溶液中,于80-100℃下反應10-40min,將所得反應液進行純化、洗滌,即得所述式(i)所示的化合物;
優選的,所述步驟(1)中,炔雌醇、式a-i所示的化合物、cuso4·5h2o、抗壞血酸鈉和三(2-苯并咪唑基甲基)胺加入的摩爾比為1∶(0.90~1.30)∶(0.18~0.22)∶(1.8~2.2)∶(0.08~0.12)。
優選的,在所述步驟(1)反應體系中,所述與水互溶性較好有機溶劑與水的體積比為1∶1。
優選的,式a-i所示的化合物如下,
上述式a-i-1所示的化合物的合成步驟包括:
q1、將式x-1所示的化合物2-(二甲氨基)溴乙烷氫溴酸鹽與疊氮化鈉混合,加水溶解,氮氣保護,87~93℃下反應15~17h,將所得反應液調節ph值至中性,將上述反應液進行tlc檢測,以二氯甲烷∶甲醇=10∶1(v/v)展開,顯色劑為碘單質,rf=0.5,采用二氯甲烷萃取,所得有機相經干燥,旋干,得油狀液體,即為式x-2所示的化合物;
q2、將式x-2所示的化合物加入醚類溶劑中溶解,氮氣保護,然后逐滴加入溴甲基硼酸頻哪醇酯的醚類溶液,滴加完畢,20-30℃繼續反應2~4h,至出現白色渾濁,用醚類溶劑沉淀,離心,真空干燥,得到化合物x-3;
q3、向式x-3所示的化合物中加入氟氫化鉀、鹽酸、水、與水互溶性較好的有機溶劑,42~47℃下反應1~3h,將上述反應液進行tlc檢測,以乙酸乙酯∶甲醇=10∶1(v/v)展開,顯色劑為碘單質,rf=0.3,用乙酸乙酯萃取,旋干,純化,得到上述式a-i-1所示的化合物;
優選的,式a-i所示的化合物如下,
上述式a-i-2所示的化合物的合成步驟包括:
f1、將式y-1所示的四聚乙二醇溶解于極性非質子性溶劑中,加入堿液,緩慢滴加含對甲苯磺酰氯的溶液,滴加完畢,置于冰浴中反應1.5~2.5h,萃取,純化,得到油狀物,即式y-2所示的化合物;
f2、將式y-2所示的化合物與疊氮化鈉溶于極性非質子性溶劑中,93~97℃下加熱反應15~17h,萃取,旋蒸,得到油狀物,即式y-3所示的化合物;
f3、將式y-3所示的化合物溶于極性非質子性溶劑中,加入4-二甲氨基吡啶和三乙胺,冰浴下攪拌,緩慢滴加含對甲苯磺酰氯的溶液,滴加完畢,室溫反應過夜,然后經洗滌、旋蒸和純化,得到黃色油狀物,即式y-4所示的化合物;
f4、將式y-4所示的化合物、n,n-二甲基乙醇胺、堿溶于極性非質子性溶劑中,67~72℃加熱反應10~12h,過濾,收集濾液,經旋蒸、萃取、旋干和純化,得式y-5所示的化合物;
f5、將式y-5所示的化合物溶于干燥的極性非質子性溶劑中,將溴甲基硼酸頻那醇酯溶于干燥的的極性非質子性溶劑中,然后將上述溴甲基硼酸頻那醇酯的溶液逐滴加入上述式y-5所示的化合物的溶液中,在室溫、氮氣保護的條件下反應2.5~3.5h,旋干,得到式y-6所示的化合物;
f6、將式y-6所示的化合物溶于與水互溶性較好的有機溶劑中,加入氟氫化鉀水溶液、鹽酸,氮氣保護,42~47℃反應1.5~2.5h,加入氨水淬滅,調節ph值至中性,旋干,純化,得到上述式a-i-2所示的化合物;
優選的,所述步驟(1)、q3、f6中,所述與水互溶性好的有機溶劑包括n,n-二甲基甲酰胺、二甲亞砜、丙酮、乙腈、甲醇和乙醇中的至少一種。
優選的,所述步驟f1中,所述極性非質子性溶劑包括四氫呋喃、n,n-二甲基甲酰胺和二氧六環中的至少一種。
優選的,所述步驟f2中,所述極性非質子性溶劑包括乙腈、二甲亞砜和n,n-二甲基甲酰胺中的至少一種。
優選的,所述步驟f3中,所述極性非質子性溶劑包括二氯甲烷、四氫呋喃、n,n-二甲基甲酰胺和二甲亞砜中至少一種。
優選的,所述步驟f4、f5中,所述極性非質子性溶劑包括四氫呋喃、n,n-二甲基甲酰胺、二甲亞砜和乙腈中的至少一種。
優選的,所述的堿液包括氫氧化鉀或氫氧化鈉。
優選的,所述的醚類溶劑包括乙醚。
本發明提供上述18f標記的炔雌醇在制備治療乳腺癌藥物中的應用。
本發明提供上述18f標記的炔雌醇在制備pet示蹤劑中的應用。
本發明還提供一種pet示蹤劑,包括上述18f標記的炔雌醇。
本發明技術方案,具有如下優點:
1.本發明在炔雌醇上引入修飾基團并經過18f標記,所獲得的18f標記的炔雌醇,具備良好的生物活性和優異的藥動學性質,而且體外穩定性好,對乳腺癌er+細胞具有較好的靶向性,可以作為pet示蹤劑,用于高轉移乳腺癌er+細胞的早期診斷或療效評價。
2.本發明還提供18f標記炔雌醇的制備方法,以炔雌醇為原料,通過click反應引入標記基團,在相對溫和的條件下加熱,通過18f-交換,簡化標記步驟和純化時間,所獲得的18f標記的炔雌醇放射化學產率高,更加有利于放射性標記化合物的商業應用于臨床推廣。
3.本發明還提供標記基團的制備方法,該制備方法反應速率快、生產效率高,條件簡單,易于工業化控制,綜合經濟效益顯著,解決了現有生產工藝中反應時間長、難以控制反應條件、無法滿足工業化生產需求的問題。
附圖說明
為了更清楚地說明本發明具體實施方式或現有技術中的技術方案,下面將對具體實施方式或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖是本發明的一些實施方式,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據這些附圖獲得其他的附圖。
圖1是本發明實施例1-6制備的18f標記的炔雌醇結構式;
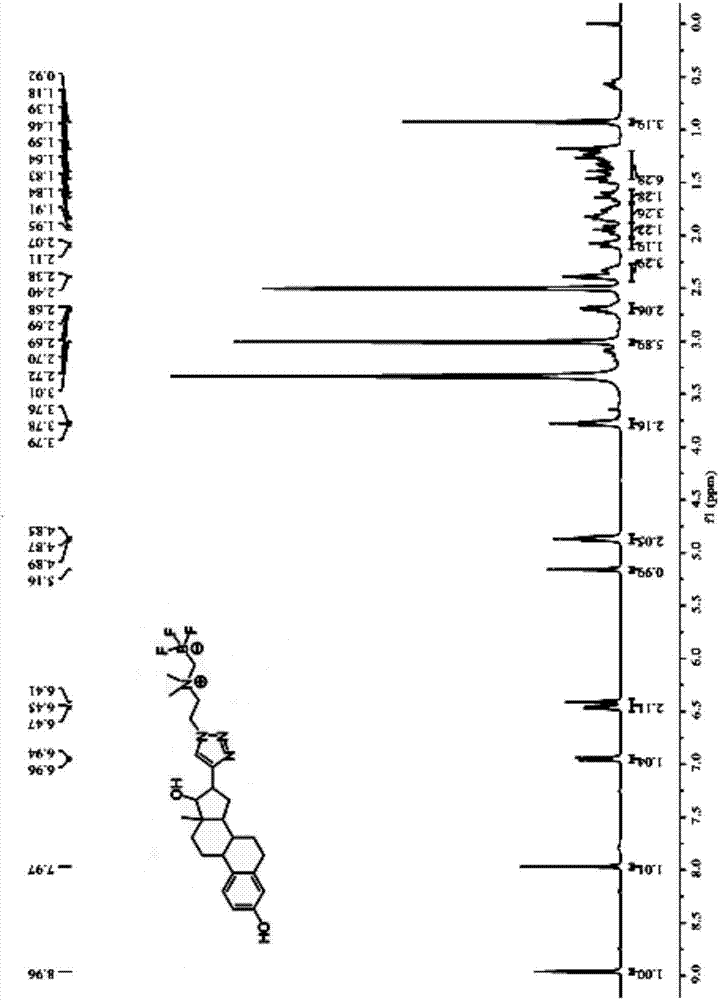
圖2是本發明實施例1制備的標記前體a-ii-a的1h-nmr圖譜;
圖3是本發明實施例4制備的標記前體a-ii-b的1h-nmr圖譜;
圖4是本發明實施例1中標記前體a-ii-a的hplc圖譜;
圖5是本發明實施例4中標記前體a-ii-b的hplc圖譜;
圖6是本發明實施例1中標記產物i-a的放射性高效液相圖譜;
圖7是本發明實施例4中標記產物i-b的放射性高效液相圖譜;
圖8是本發明實施例1中標記前體a-ii-a不同溫度下的標記率;
圖9是本發明實施例4中標記前體a-ii-b不同溫度下的標記率;
圖10是本發明實施例1中標記前體a-ii-a體外穩定性測試圖譜;
圖11是本發明實施例4中標記前體a-ii-b體外穩定性測試圖譜;
圖12是本發明實施例1中標記產物i-a在pbs中孵育4h后radio-hplc分析圖譜;
圖13是本發明實施例1中標記產物i-a在血清中孵育4h后radio-hplc分析圖譜;
圖14是本發明實施例1中標記產物i-a在pbs和血清中孵育不同時間的放化純圖譜;
圖15是本發明實施例4中標記產物i-b在pbs中孵育4h后radio-hplc分析圖譜;
圖16是本發明實施例4中標記產物i-b在血清中孵育4h后radio-hplc分析圖譜;
圖17是本發明實施例4中標記產物i-b在pbs和血清中孵育不同時間的放化純圖譜;
圖18是在本發明實施例1中不同濃度標記前體a-ii-a下t47d細胞生存率;
圖19是在本發明實施例4中不同濃度標記前體a-ii-b下t47d細胞生存率;
圖20是本發明實施例1中標記產物i-a在t47d細胞中的總攝取率與阻斷組攝取率;
圖21是本發明實施例4中標記產物i-b在t47d細胞中的總攝取率與阻斷組攝取率。
具體實施方式
下面將結合附圖對本發明的技術方案進行清楚、完整地描述,顯然,所描述的實施例是本發明一部分實施例,而不是全部的實施例。基于本發明中的實施例,本領域普通技術人員在沒有做出創造性勞動前提下所獲得的所有其他實施例,都屬于本發明保護的范圍。此外,下面所描述的本發明不同實施方式中所涉及的技術特征只要彼此之間未構成沖突就可以相互結合。
下述實施例中涉及到的試劑如下:
2-(二甲氨基)溴乙烷氫溴酸鹽、疊氮化鈉、溴甲基硼酸頻哪醇酯、氟氫化鉀、四聚乙二醇、對甲苯磺酰氯、n,n-二甲基乙醇胺、乙醚、乙酸乙酯、n,n-二甲基甲酰胺、cuso4·5h2o、naasc、三(2-苯并咪唑基甲基)胺、四氫呋喃、二氯甲烷、甲醇、乙醇、正己烷、鹽酸、氯化鈉。所有試劑均為分析純,使用前未經純化,購置于阿拉丁試劑公司。
炔雌醇的生產廠家為安耐吉化學。
下述實施例中涉及到的儀器如下:
核磁共振儀(brukerdrx-500)、高效液相色譜儀(waters2445),γ計數儀(perkin-elmer1470)。
實施例1
本實施例所述的具有式(i)結構18f標記的炔雌醇,如圖1所示,其中r1為亞乙基,具有式(i-a)結構:
化合物(i-a)的合成路線為:
上述式i-a所示的化合物的合成步驟包括:
q1、將式x-1所示的化合物2-(二甲氨基)溴乙烷氫溴酸鹽2.33g(10mmol)與疊氮化鈉2.60g(40mmol)混合,加水50ml溶解,氮氣保護,90℃下反應16h,反應結束加入na2co3將所得反應液調節ph值至中性,將上述反應液進行tlc檢測,以二氯甲烷∶甲醇=10∶1(v/v)展開,顯色劑為碘單質,rf=0.5,采用二氯甲烷萃取,所得有機相經無水硫酸鈉干燥,旋干,得油狀液體,質譜檢測:115為m+h+,即為式x-2所示的化合物;
q2、將式x-2所示的化合物757mg(6.63mmol)加入10ml乙醚溶解,氮氣保護,然后逐滴加入含有溴甲基硼酸頻哪醇酯1537.8mg(6.96mmol)的乙醚溶液5ml,滴加完畢,25℃繼續反應3h,至出現白色渾濁,加入乙醚20ml繼續沉淀,離心,40℃真空干燥,質譜檢測m+na:255.,得到化合物x-3;
q3、向式x-3所示的化合物1.73g(6.78mmol)中加入氟氫化鉀溶液(3m)4ml、鹽酸(4m)4ml、水2ml、n,n-二甲基甲酰胺2ml,45℃下反應2h,將上述反應液進行tlc檢測,以乙酸乙酯∶甲醇=10∶1(v/v)展開,顯色劑為碘單質,rf=0.3,用乙酸乙酯萃取,旋干,經硅膠柱層析純化,質譜檢測:177(m-f-),219(m+na+),415(2m+na+),得到上述式a-i-1所示的化合物;
q4、取炔雌醇50.4mg(0.17mmol)溶解于1.5ml的n,n-二甲基甲酰胺中,將式a-i-1所示的化合物40.5mg(0.22mmol)加入上述的炔雌醇溶液中,然后加入cuso4·5h2o8.44mg(0.034mmol)的水溶液300μl、naasc66.8mg(0.34mmol)的水溶液1000μl和三(2-苯并咪唑基甲基)胺7.2mg(0.017mmol)的水溶液200μl,在45℃下反應2h,將所得反應液旋干,加入n,n-二甲基甲酰胺30ml并離心,收集上清液,即得式a-ii-a所示的化合物;
q5、將所述式a-ii-a所示的化合物1.23mg(0.0025mmol)溶于n,n-二甲基甲酰胺100μl中,配制成25nm/μl溶液,然后用300μl噠嗪-鹽酸-n,n-二甲基甲酰胺(ph=2.5)緩沖液將200mci的18f-溶液經qma柱洗脫,收集,取20μl式a-ii-a所示的化合物加入到收集的18f-溶液中,在90℃下反應20min,將所得反應液用水稀釋,然后負載c18柱純化,用水洗滌三次,每次10ml,經c18柱純化后,放化純>98%,即得所述式(i-a)所示的化合物。
將上述制備得到的標記前體a-ii-a進行1h-nmr分析,其圖譜如圖2所示。
實施例2
本實施例所述的具有式(i)所示結構18f標記的炔雌醇,如圖1所示,,其中r1為亞乙基,具有式(i-a)結構:
化合物(i-a)的合成路線為:
上述式i-a所示的化合物的合成步驟包括:
q1、將式x-1所示的化合物2-(二甲氨基)溴乙烷氫溴酸鹽4.66g(20mmol)與疊氮化鈉4.9g(76mmol)混合,加水100ml溶解,氮氣保護,93℃下反應15h,反應結束加入na2co3將所得反應液調節ph值至中性,將上述反應液進行tlc檢測,以二氯甲烷∶甲醇=10∶1(v/v)展開,顯色劑為碘單質,rf=0.5,采用二氯甲烷萃取,所得有機相經無水硫酸鈉干燥,旋干,得油狀液體,即為式x-2所示的化合物;
q2、將式x-2所示的化合物1484.0mg(13mmol)加入20ml乙醚溶解,氮氣保護,然后逐滴加入溴甲基硼酸頻哪醇酯3158.9mg(14.3mmol)的乙醚溶液10ml,滴加完畢,20℃繼續反應4h,至出現白色渾濁,加入乙醚40ml繼續沉淀,離心,42℃真空干燥,得到化合物x-3;
q3、向式x-3所示的化合物2.552g(10mmol)中加入氟氫化鉀(3m)6ml、鹽酸(4m)6ml、水3ml、二甲亞砜3ml,42℃下反應3h,將上述反應液進行tlc檢測,以乙酸乙酯∶甲醇=10∶1(v/v)展開,顯色劑為碘單質,rf=0.3,用乙酸乙酯萃取,旋干,經硅膠柱層析純化,得到上述式a-i-1所示的化合物;
q4、取炔雌醇118.5mg(0.4mmol)溶解于4ml的甲醇中,將式a-i-1所示的化合物81.0mg(0.44mmol)加入上述的炔雌醇溶液中,然后加入cuso4·5h2o22.0mg(0.088mmol)的水溶液600μl、naasc142.6mg(0.72mmol)的水溶液2000μl和三(2-苯并咪唑基甲基)胺20.33mg(0.048mmol)的水溶液400μl,控制上述反應體系中甲醇與h2o體積比為1∶1.1,在40℃下反應1.5h,將所得反應液旋干,加入甲醇溶液70ml并離心,收集上清液,即得式a-ii-a所示的化合物;
q5、將所述式a-ii-a所示的化合物2.26mg(0.0050mmol)溶于n,n-二甲基甲酰胺200ml中,配制成25nm/μl溶液,然后用500μl噠嗪-鹽酸-n,n-二甲基甲酰胺(ph=2.5)緩沖液將200mci的18f-溶液經qma柱洗脫,收集,取20μl式a-ii-a所示的化合物加入到收集的18f-溶液中,在90℃下反應20min,將所得反應液用水稀釋,然后負載c18柱純化,用水洗滌三次,每次10ml,即得所述式(i-a)所示的化合物。
實施例3
本實施例所述的具有式(i)所示結構18f標記的炔雌醇,如圖1所示,其中r1為亞乙基,具有式(i-a)結構:
化合物(i-a)的合成路線為:
上述式i-a所示的化合物的合成步驟包括:
q1、將式x-1所示的化合物2-(二甲氨基)溴乙烷氫溴酸鹽3493.5mg(15mmol)與疊氮化鈉4095.6mg(63mmol)混合,加水75ml溶解,氮氣保護,87℃下反應17h,反應結束加入na2co3將所得反應液調節ph值至中性,將上述反應液進行tlc檢測,以二氯甲烷∶甲醇=10∶1(v/v)展開,顯色劑為碘單質,rf=0.5,采用二氯甲烷萃取,所得有機相經無水硫酸鈉干燥,旋干,得油狀液體,即為式x-2所示的化合物;
q2、將式x-2所示的化合物1141.5mg(10mmol)加入15ml乙醚溶解,氮氣保護,然后逐滴加入溴甲基硼酸頻哪醇酯3556.5mg(16.1mmol)的乙醚溶液5ml,滴加完畢,30℃繼續反應2h,至出現白色渾濁,加入乙醚30ml繼續沉淀,離心,41℃真空干燥,得到化合物x-3;
q3、向式x-3所示的化合物2041.6mg(8mmol)中加入氟氫化鉀(3m)5ml、鹽酸(4m)5ml、水2.5ml、乙腈2.5ml,47℃下反應1h,將上述反應液進行tlc檢測,以乙酸乙酯∶甲醇=10∶1(v/v)展開,顯色劑碘單質,rf=0.3,用乙酸乙酯萃取,旋干,經硅膠柱層析純化,得到上述式a-i-1所示的化合物;
q4、取炔雌醇88.9mg(0.3mmol)溶解于2.2ml二甲亞砜中,將式a-i-1所示的化合物49.7mg(0.27mmol)加入上述的炔雌醇溶液中,然后加入cuso4·5h2o13.5mg(0.054mmol)的水溶液450μl、naasc107.0mg(0.54mmol)的水溶液3000μl和三(2-苯并咪唑基甲基)胺9.8mg(0.024mmol)的水溶液300μl,控制上述反應體系中二甲亞砜與h2o體積比為1∶1,在50℃下反應0.5h,將所得反應液旋干,加入二甲亞砜45ml并離心,收集上清液,即得式a-ii-a所示的化合物;
q5、將所述式a-ii-a所示的化合物3.69mg(0.0075mmol)溶于n,n-二甲基甲酰胺300ml中,配制成25nm/μl溶液,然后用900μl噠嗪-鹽酸-n,n-二甲基甲酰胺(ph=2.5)緩沖液將600mci的18f-溶液經qma柱洗脫,收集,取20μl式a-ii-a所示的化合物加入到收集的18f-溶液中,在90℃下反應20min,將所得反應液用水稀釋,然后負載c18柱純化,用水洗滌三次,每次10ml,即得所述式(1-a)所示的化合物。
實施例4
本實施例所述的具有式(i)所示結構18f標記的炔雌醇,如圖1所示,其中r1為3,6,9,12-四氧雜十四烷亞基,具有式(i-b)結構:
化合物(i-b)的合成路線為:
上述式i-a所示的化合物的合成步驟包括:
f1、室溫下,將式y-1所示的四聚乙二醇4464.8mg(23mmol)溶解20ml四氫呋喃中,加入koh2917.5mg(52mmol)水溶液3ml,緩慢滴加含對甲苯磺酰氯4385.0mg(23mmol)的四氫呋喃溶液30ml,滴加完畢,置于冰浴中反應2h,反應完畢采用二氯甲烷萃取,經柱層析分離純化(二氯甲烷/甲醇=30/1,v/v),得到油狀物,即式y-2所示的化合物;
f2、將式y-2所示的化合物3481.2mg(10mmol)與疊氮化鈉1800.0mg(30mmol)溶于20ml乙腈中,在95℃下加熱反應16h,反應結束采用二氯甲烷萃取,旋蒸,得到油狀物,即式y-3所示的化合物;
f3、將式y-3所示的化合物1468.1g(6.7mmol)溶于20ml二氯甲烷中,加入4-二甲氨基吡啶357.2mg(2.9mmol)和三乙胺8.9ml(67mmol),冰浴下攪拌,緩慢滴加含對甲苯磺酰氯3813.0g(20mmol)的二氯甲烷溶液20ml,滴加完畢,室溫反應過夜,分別用1m鹽酸和飽和食鹽水洗滌各洗滌三次,有機相旋蒸,經柱層析分離純化(正己烷/乙酸乙酯=2/1,v/v),得到黃色油狀物,即式y-4所示的化合物;
f4、將式y-4所示的化合物3208.9g(8.6mmol)、n,n-二甲基乙醇胺772μl(668.1mg,7.5mmol)、koh1627.1mg(29mmol)溶于50ml四氫呋喃中,在70℃加熱反應12h,反應結束后過濾,收集濾液,旋蒸,用二氯甲烷萃取,有機相旋干,經柱層析分離純化(二氯甲烷/甲醇=10/1,v/v),得式y-5所示的化合物;
f5、將式y-5所示的化合物480mg(1.6mmol)溶于10ml干燥的四氫呋喃中,將溴甲基硼酸頻那醇酯279.4μl(352.0mg,1.6mmol)溶于5ml干燥的四氫呋喃中,然后將上述溴甲基硼酸頻那醇酯的四氫呋喃溶液逐滴加入上述式y-5所示的化合物的四氫呋喃溶液中,在室溫、氮氣保護的條件下反應3h,將四氫呋喃旋干,得到式y-6所示的化合物;
f6、將式y-6所示的化合物478mg(1.1mmol)溶于1mln,n-二甲基甲酰胺中,加入1ml氟氫化鉀水溶液(4m)、1ml鹽酸(3m),氮氣保護,在45℃反應2h,加入10μl氨水淬滅,調節ph值至中性,旋干,經柱層析分離純化(二氯甲烷/甲醇=20/1,v/v),得到上述式a-i-2所示的化合物;
f7、取炔雌醇53.3mg(0.18mmol)溶解于4mln,n-二甲基甲酰胺/水(1/1,v/v)中,將式a-i-2所示的化合物66mg(0.18mmol)加入上述的炔雌醇溶液中,然后加入cuso4·5h2o9mg(0.036mmol)、naasc71.3mg(0.36mmol)和三(2-苯并咪唑基甲基)胺7.2mg(0.018mmol),控制上述反應體系中n,n-二甲基甲酰胺與h2o體積比為1∶0.9,在45℃下反應2h,將所得反應液旋干,加入n,n-二甲基甲酰胺30ml并離心,取上清液,即得式a-ii-b所示的化合物;
f8、將所述式a-ii-b所示的化合物1.67mg(0.0025mmol)溶于n,n-二甲基甲酰胺100μl中,配制成25nm/μl溶液,然后用300μl噠嗪-鹽酸-n,n-二甲基甲酰胺(ph=2.5)緩沖液將200mci的18f-溶液經qma柱洗脫,收集,取20μl式a-ii-b所示的化合物加入到收集的18f-溶液中,在85℃下反應30min,用水稀釋,然后負載c18柱純化,用水洗滌三次,每次10ml,即得所述式(i-a)所示的化合物。
將上述制備的標記前體a-ii-b進行1h-nmr分析,其圖譜分別如圖3所示。
實施例5
本實施例所述的具有式(i)所示結構18f標記的炔雌醇,如圖1所示,其中r1為3,6,9,12-四氧雜十四烷亞基,具有式(i-b)結構:
化合物(i-b)的合成路線為:
上述式i-a所示的化合物的合成步驟包括:
f1、室溫下,將式y-1所示的四聚乙二醇9076mg(50mmol)溶解于40ml二氧六環中,加入naoh4001mg(100mmol)水溶液6ml,緩慢滴加含對甲苯磺酰氯9532.5mg(50mmol)的二氧六環溶液60ml,滴加完畢,置于冰浴中反應2.5h,反應完畢采用二氯甲烷萃取,經柱層析分離純化(二氯甲烷/甲醇=30/1,v/v),旋干,得到油狀物,即式y-2所示的化合物;
f2、將式y-2所示的化合物5221.8mg(15mmol)與疊氮化鈉2700.5mg(45mmol)溶于30ml二甲亞砜中,在97℃下加熱反應17h,反應結束采用二氯甲烷萃取,旋蒸,得到油狀物,即式y-3所示的化合物;
f3、將式y-3所示的化合物1095.6mg(5mmol)溶于15ml乙腈中,加入4-二甲氨基吡啶258.7mg(2.1mmol)和三乙胺6.6ml(50mmol),冰浴下攪拌,緩慢滴加含對甲苯磺酰氯2859.8mg(15mmol)的乙腈溶液15ml,滴加完畢,室溫反應過夜,分別用1m鹽酸和飽和食鹽水洗滌各洗滌三次,有機相旋蒸,經柱層析分離純化(正己烷/乙酸乙酯=2/1,v/v),得到黃色油狀物,即式y-4所示的化合物;
f4、將式y-4所示的化合物2238.8mg(6mmol)、n,n-二甲基乙醇胺535.3μl(463.2mg,5.2mmol)、koh1133.4mg(20.2mmol)溶于35ml四氫呋喃中,在65℃下加熱反應12h,反應結束后過濾,收集濾液,旋蒸,用二氯甲烷萃取,有機相旋干,經柱層析分離純化(二氯甲烷/甲醇=10/1,v/v),得式y-5所示的化合物;
f5、將式y-5所示的化合物300mg(1mmol)溶于5ml干燥的乙腈中,將溴甲基硼酸頻那醇酯174.6μl(220mg,1mmol)溶于2.5ml干燥的乙腈中,然后將上述溴甲基硼酸頻那醇酯的乙腈溶液逐滴加入上述式y-5所示的化合物的乙腈溶液中,在室溫、氮氣保護的條件下反應3.5h,將乙腈旋干,得到式y-6所示的化合物;
f6、將式y-6所示的化合物217.3mg(0.5mmol)溶于0.5ml乙醇中,加入0.5ml氟氫化鉀水溶液(4m)、0.5ml鹽酸(3m),氮氣保護,在47℃反應1.5h,加入5μl氨水淬滅,調節ph值至中性,旋干,經柱層析分離純化(二氯甲烷/甲醇=20/1,v/v),得到上述式a-i-2所示的化合物;
f7、取炔雌醇44.4mg(0.15mmol)溶解于4ml丙酮/水(1/1,v/v)中,將式a-i-2所示的化合物71.5mg(0.195mmol)加入上述的炔雌醇溶液中,然后加入cuso4·5h2o6.75mg(0.027mmol)、naasc53.5mg(0.27mmol)和三(2-苯并咪唑基甲基)胺4.9mg(0.12mmol),控制上述反應體系中丙酮與h2o體積比為1∶1,在50℃下反應0.5h,將所得反應液旋干,加入丙酮25ml并離心,取上清液,即得式a-ii-b所示的化合物;
f8、將所述式a-ii-b所示的化合物1.34mg(0.0020mmol)溶于n,n-二甲基甲酰胺80μl中,配制成25nm/μl溶液,然后用240μl噠嗪-鹽酸-n,n-二甲基甲酰胺(ph=2.5)緩沖液將160mci的18f-溶液經qma柱洗脫,收集,取20μl式a-ii-b所示的化合物加入到收集的18f-溶液中,在80℃下反應30min,用水稀釋,然后負載c18柱純化,用水洗滌三次,每次10ml,即得所述式(i-b)所示的化合物。
實施例6
本實施例所述的具有式(i)所示結構18f標記的炔雌醇,如圖1所示,其中r1為3,6,9,12-四氧雜十四烷亞基,具有式(i-b)結構:
化合物(i-b)的合成路線為:
上述式i-a所示的化合物的合成步驟包括:
f1、室溫下,將式y-1所示的四聚乙二醇1941.2mg(10mmol)溶解10mln,n-二甲基甲酰胺中,加入koh1268.5mg(22.6mmol)水溶液1.5ml,緩慢滴加含對甲苯磺酰氯1906.5mg(10mmol)的n,n-二甲基甲酰胺溶液15ml,滴加完畢,置于冰浴中反應1.5h,反應完畢采用二氯甲烷萃取,經柱層析分離純化(二氯甲烷/甲醇=30/1,v/v),旋干,得到油狀物,即式y-2所示的化合物;
f2、將式y-2所示的化合物1740.6mg(5mmol)與疊氮化鈉900.0mg(15mmol)溶于10mln,n-二甲基甲酰胺中,在93℃下加熱反應15h,反應結束采用二氯甲烷萃取,旋蒸,得到油狀物,即式y-3所示的化合物;
f3、將式y-3所示的化合物657.4mg(3mmol)溶于10ml二甲亞砜中,加入4-二甲氨基吡啶160.1g(1.3mmol)和三乙胺4.0ml(30mmol),冰浴下攪拌,緩慢滴加含對甲苯磺酰氯1715.9mg(9.0mmol)的二甲亞砜溶液10ml,滴加完畢,室溫反應過夜,分別用1m鹽酸和飽和食鹽水洗滌各洗滌三次,有機相旋蒸,經柱層析分離純化(正己烷/乙酸乙酯=2/1,v/v),得到黃色油狀物,即式y-4所示的化合物;
f4、將式y-4所示的化合物1492.5mg(4mmol)、n,n-二甲基乙醇胺360.3μl(311.8mg,3.5mmol)、koh756.8mg(13.5mmol)溶于25ml二甲亞砜中,在67℃加熱反應10h,反應結束后過濾,收集濾液,旋蒸,用二氯甲烷萃取,有機相旋干,經柱層析分離純化(二氯甲烷/甲醇=10/1,v/v),得式y-5所示的化合物;
f5、將式y-5所示的化合物900mg(3mmol)溶于20ml干燥的n,n-二甲基甲酰胺中,將溴甲基硼酸頻那醇酯523.9μl(660mg,3mmol)溶于10ml干燥的的n,n-二甲基甲酰胺中,然后將上述溴甲基硼酸頻那醇酯的n,n-二甲基甲酰胺溶液逐滴加入上述式y-5所示的化合物的n,n-二甲基甲酰胺溶液中,在室溫、氮氣保護的條件下反應3h,將n,n-二甲基甲酰胺旋干,得到式y-6所示的化合物;
f6、將式y-6所示的化合物869.2mg(2.0mmol)溶于2ml乙腈中,加入2ml氟氫化鉀水溶液(4m)、2ml鹽酸(3m),氮氣保護,在42℃反應1.5h,加入20μl氨水淬滅,調節ph值至中性,旋干,經柱層析分離純化(二氯甲烷/甲醇=20/1,v/v),得到上述式a-i-2所示的化合物;
f7、取炔雌醇59.2mg(0.2mmol)溶解于5ml乙腈/水(1/1,v/v)中,將式a-i-2所示的化合物66.0mg(0.18mmol)加入上述的炔雌醇溶液中,然后加入cuso4·5h2o11.0mg(0.044mmol)、naasc87.2mg(0.044mmol)和三(2-苯并咪唑基甲基)胺9.8mg(0.024mmol),控制上述反應體系中乙腈與h2o體積比為1∶1.1,在40℃下反應1.0h,將所得反應液旋干,加入乙腈30ml并離心,取上清液,即得式a-ii-b所示的化合物;
f8、將所述式a-ii-b所示的化合物2.01mg(0.0030mmol)溶于n,n-二甲基甲酰胺120μl中,配制成25nm/μl溶液,然后用450μl噠嗪-鹽酸-n,n-二甲基甲酰胺(ph=2.5)緩沖液將300mci的18f-溶液經qma柱洗脫,收集,取20μl式a-ii-b所示的化合物加入到收集的18f-溶液中,在85℃下反應30min,用水稀釋,然后負載c18柱純化,用水洗滌三次,每次15ml,即得所述式(i-b)所示的化合物。
實驗例
一、標記前體和產物的純度分析
1.標記前體的純度分析
在280nm紫外波長下,將實施例1制備的標記前體a-ii-a、實施例4制備的標記前體a-ii-b分別進行液相分析,結果如圖4、圖5所示,采用實施例1標記前體a-ii-a、實施例4制備標記前體a-ii-b的高效液相檢測顯示均為一單峰。其中,標記前體a-ii-a純度為99%,保留時間tr為16.1min;標記前體a-ii-b純度為99%,保留時間tr為16.5min。
2.標記產物的純度分析
在280nm紫外波長下,將實施例1制備的標記產物i-a、實施例4制備的標記產物i-b分別進行放射性高效液相分析,結果如圖6、圖7所示,其中圖6為標記產物i-a純化后radio-hplc分析圖譜,圖7為標記產物i-b純化后radio-hplc分析圖譜,采用實施例1制備的標記產物i-a、實施例4制備的標記產物i-b的放射性高效液相檢測顯示均為一單峰。其中,標記產物i-a純度為99%,保留時間tr為16.3min;標記產物i-b純度為98%,保留時間tr為16.6min。
將實施例1制備的標記前體a-ii-a、實施例4制備的標記前體a-ii-b分別溶于dmf溶液,配制25nm/μl的溶液。采用200mci的18f-用300μl的pry-hcl-dmf(ph=2.5)的緩沖液從qma柱上洗脫下來,分別用ep管收集,將標記前體a-ii-a、a-ii-b的dmf溶液分別加入到上述含18f-的ep管中,不同溫度下分別加熱20min。反應混合物用水稀釋后負載到c18柱上,用水(3×10m1)洗滌。純的放射性標記產物用乙醇與水(1∶1)洗脫到小玻璃瓶中。取一點樣品通過hplc進行質量控制分析。所得標記率如圖8、圖9所示。80℃是一個有效標記溫度并用于后續實驗。經c18柱純化后,放化純>98%。
二、體外穩定性測試
1.標記前體的體外穩定性測試
將實施例1制備的標記前體a-ii-a、實施例4制備的標記前體a-ii-b分別與噠嗪-鹽酸(ph2.0-2.5)的緩沖液一起在不同溫度下加熱20min,用hplc檢測其純度,結果如圖10、圖11所示。
2.標記產物的體外穩定性測試
取100μci放射性的實施例1制備的標記產物i-a、實施例4制備的標記產物i-b分別加入到500μlpbs或血清(該血清biologicalindustries提供)中,并37℃分別孵育0,1,2,4h。在預定時間點,取出pbs混合液中一部分,直接進行hplc分析;對于血清樣品,取70%mecn加至血清中,10000g離心5min,取上清用hplc分析。結果顯示,在4h后,標記產物i-a的放化純仍大于97%。其中,圖12為標記產物i-a在pbs中孵育4h后radio-hplc分析圖譜,圖13為標記產物i-a在血清中孵育4h后radio-hplc分析圖譜,圖14為標記產物i-a在pbs和血清中孵育不同時間的放化純圖譜;圖15為標記產物i-b在pbs中孵育4h后radio-hplc分析圖譜,圖16為標記產物i-b在血清中孵育4h后radio-hplc分析圖譜,圖17為標記產物i-b在pbs和血清中孵育不同時間的放化純圖譜。
三、脂水分配系數
1.標記產物i-a脂水分配系數測定
將pbs緩沖液與等體積的正辛醇充分振蕩,使兩相相互飽和,室溫下靜置24h以上,用分液漏斗將兩相分離。取實施例1制備的標記產物i-a溶液0.05ml,加入1ml正辛醇、0.95mlpbs緩沖液混合,用渦旋混合器振蕩5min,充分混勻,然后4000g離心5min,在有機相和水相中分別取100μl樣品于γ計數管中,用γ計數儀測定其放射性活度,實驗重復兩次。計算脂水分配系數的logp值。用公式logp=log(cpm水相/cpm有機相)計算,實際得到:logp=0.52±0.09,為親脂性。
2.標記產物i-b脂水分配系數測定
將pbs緩沖液與等體積的正辛醇充分振蕩,使兩相相互飽和,室溫下靜置24h以上,用分液漏斗將兩相分離。取實施例2制備的標記產物i-b溶液0.05ml,加入1ml正辛醇、0.95mlpbs緩沖液混合,用渦旋混合器振蕩5min,充分混勻,然后4000g離心5min,在有機相和水相中分別取100μl樣品于γ計數管中,用γ計數儀測定其放射性活度,實驗重復兩次。計算脂水分配系數的logp值。用公式logp=log(cpm水相/cpm有機相)計算,實際得到:logp=-0.176±0.028,為親水性。
四、細胞毒性實驗
以6000個細胞/孔的密度將t47d細胞(所述細胞由南京科佰生物科技有限公司提供)接種到96孔板中,培養基體積100μl/孔,37℃過夜培養12h,吸除原有培養基后每孔加入200μl的含藥濃度為12.5、25、50、100μm培養基,每個濃度設置至少6個復孔,然后分別放入培養箱分別培養3h,6h,12h,24h,達到培養時間時,每孔加入20μl的mtt(5mg/mlpbs)溶液,放入培養箱中繼續培養4h后,小心吸除上清液后,每孔加入150μl的dmso,放置在搖床上震蕩至孔底紫色結晶完全溶解,用酶標儀測定490nm波長下的吸光度od值,然后計算細胞生存率。標記前體a-ii-a、a-ii-b細胞毒性實驗結果分別如圖18、19所示,在100μm藥物濃度下,t47d細胞生存率分別為89.2%、88.9%,說明標記前體a-ii-a、a-ii-b對細胞是無毒的。
五、細胞攝取與阻斷
1.標記產物i-a
將t47d乳腺癌細胞接種在24孔板,密度為2×105細胞/孔,過夜培養。t47d細胞由實施例1制備的標記產物i-a在37℃下進行培養15,30,60,120min,每孔0.5μci。在阻斷特異性結合的實驗中,將雌二醇加入到24孔板的t47d細胞中,終濃度5μm。培養結束后,t47d細胞用pbs洗三遍,用0.1m的naoh裂解,收集裂解液,保留的放射性采用γ計數器測量。細胞攝取采用加入的劑量百分率表述(%ad),該劑量經衰變校正。所有的實驗均三個復孔重復兩次。
實驗結果如圖20所示,細胞總攝取率為3.5%,并能夠被雌二醇阻斷至1.7%,證明此標記產物i-a對t47d細胞具有靶向性。
2.標記產物i-b
將t47d乳腺癌細胞接種在24孔板,密度為2×105細胞/孔,過夜培養。t47d細胞由實施例4制備的標記產物i-b在37℃下進行培養15,30,60,120min,每孔0.5μci。在阻斷特異性結合的實驗中,將雌二醇加入到24孔板的t47d細胞中,終濃度5μm。培養結束后,t47d細胞用pbs洗三遍,用0.1m的naoh裂解,收集裂解液,保留的放射性采用γ計數器測量。細胞攝取采用加入的劑量百分率表述(%ad),該劑量經衰變校正。所有的實驗均三個復孔重復兩次。
實驗結果如圖21所示,細胞總攝取率為1.61%,并能夠被雌二醇阻斷至0.94%,證明此標記產物i-b對t47d細胞具有靶向性。
顯然,上述實施例僅僅是為清楚地說明所作的舉例,而并非對實施方式的限定。對于所屬領域的普通技術人員來說,在上述說明的基礎上還可以做出其它不同形式的變化或變動。這里無需也無法對所有的實施方式予以窮舉。而由此所引伸出的顯而易見的變化或變動仍處于本發明創造的保護范圍之中。
- 還沒有人留言評論。精彩留言會獲得點贊!