一種光引發的活性自由基聚合方法與流程
本發明涉及一種活性自由基聚合的方法,尤其涉及一種室溫下光引發的苯甲醛衍生物/raft試劑調控的活性自由基聚合方法。使用該引發體系,可以實現(甲基)丙烯酸酯類單體的光引發活性自由基聚合。
背景技術:
上世紀九十年代,由自由基聚合與活性聚合相結合而發展起來的“活性”/可控自由基聚合(lrp),經過二十多年的發展,lrp已被廣泛應用于高分子合成領域。在發展較成熟的lrp中,可逆加成-斷裂鏈轉移自由基(raft)聚合和原子轉移自由基聚合(atrp)具有可控性好、單體適用性強和分子設計能力強的優點。
raft聚合的引發階段與普通自由基聚合相同,必須在體系中引入自由基。目前raft聚合的引發方式主要包括引發劑的熱分解、紫外輻照、γ射線輻照、等離子體引發以及單體自身熱引發等類型。其中,光引發由于具有低能耗、高效率、經濟和環保等優點,近年來受到了廣泛關注。相比于傳統的熱引發聚合,光引發聚合具有明顯的優勢:其聚合溫度可以降到室溫甚至低于室溫,這使得單體發生副反應的幾率降低,例如鏈轉移反應。因此,特別適用于溫敏性單體的聚合。另外,光聚合可用于較低聚合上限溫度的單體的聚合中,擴大了單體的適用范圍。因此,光聚合具有巨大的應用前景。
一些金屬(如ru,ir,mn和zn等)的絡合物光敏劑或催化劑已經被應用于光激發的raft聚合中。金屬催化劑催化的raft聚合,能在室溫下使苯乙烯、(甲基)丙烯酸酯和(甲基)丙烯酰胺等單體有效聚合,并且擁有較窄的相對分子量分布,即聚合反應本身的可控性較好。但由于聚合產物中殘留的金屬催化劑不易從產品中完全去除,在使用過程中可能會導致制品的老化失效。并且由于金屬價格昂貴,不僅增加了成本,甚至有些金屬還有毒性,由此阻礙了金屬催化劑在生物藥用合成和食品包裝等方面的應用。因此,研究人員便開發出了無金屬有機光氧化還原催化劑,用于光激發的raft聚合。例如chen等人用10-苯基吩噻嗪為有機光氧化還原催化劑,以raft試劑為調控劑,在可見光照射下成功實現了丙烯酸酯及丙烯酰胺單體的可控聚合(參見chenm,macleodmj,johnsonja.[j].acsmacroletters,2015,4(5):566-569)。但由于吩噻嗪類衍生物的結構復雜,成本較高,因此,開發成本較低的有機光催化劑用于raft聚合體系,并且實現室溫下的光引發聚合,探索更加溫和、更加綠色以及更加節能的新的聚合方法,具有重要的意義。
技術實現要素:
有鑒于此,本發明的目的在于提供一種新的“活性”/可控自由基聚合的方法和綠色反應工藝。在室溫及光照的條件下,以苯甲醛衍生物/raft試劑為催化體系,催化單體聚合,實現聚合物分子量隨轉化率的增加而增加,同時達到控制相對分子量的目的。并保證所得的聚合物具有再引發活性,可用來引發第二單體的聚合,從而制備嵌段共聚物。
為實現上述目的,本發明提供如下技術方案:
一種室溫下光引發的活性自由基聚合方法,其特征在于:所述反應體系包括單體、催化劑、raft試劑;其中,按摩爾比,[單體]:[raft試劑]:[催化劑]=(150~200):1:(40~833),反應溫度為室溫,光照強度為800μwcm-2,反應時間為48~168h;催化劑為苯甲醛衍生物,單體包括甲基丙烯酸甲酯、甲基丙烯酸芐酯、甲基丙烯酸羥丙酯、聚乙二醇甲基丙烯酸酯或甲基丙烯酸n,n-二甲氨基乙酯一種或多種。
單體當以溶液加入時,單體質量濃度在20%~24%范圍內。
配置反應體系,在室溫(15℃~25℃)及光照(例如采用23w家用緊湊型節能燈)條件下進行聚合反應,制備得到聚合物,所述反應體系包括單體、催化劑、raft試劑和溶劑(不是必須的反應物);其中,[單體]:[raft試劑]:[催化劑]的摩爾比=(150~200):1:(40~833),單體濃度在20%~24%范圍內,反應溫度為室溫,反應時間為48~168h。
所述單體包括甲基丙烯酸酯類單體,例如甲基丙烯酸甲酯(mma)、甲基丙烯酸芐酯(bnma)、甲基丙烯酸羥丙酯(hpma)、聚乙二醇甲基丙烯酸酯(pegma)和甲基丙烯酸n,n-二甲氨基乙酯(dmaema)。
所述raft試劑包括但不限于4-氰基-4-(苯基硫代甲酰硫基)戊酸(ctp)、二硫代苯甲酸芐酯(bdb)、二硫代苯甲酸異丙苯酯(cdb)和s-正十二烷基-s'-(2-甲基-2-丙酸基)三硫代碳酸酯(ddmat)。
所述催化劑為苯甲醛的衍生物,包括但不限于2,4-二甲氧基苯甲醛、對甲氧基苯甲醛、對氰基苯甲醛和對二甲氨基苯甲醛。
所述溶劑為n,n-二甲基甲酰胺(dmf)、乙腈、苯甲醚和四氫呋喃(thf)等,也可以采用這些溶劑的混合物。
聚合的實施方法需要排除反應物和反應器中的氧。將上述單體、催化劑、raft試劑和溶劑按比例依次加入反應容器中充分溶解搖勻,密封。經三次冷凍抽排除氧,最后通入惰性氣體,使聚合反應在惰性氣氛中進行。最后在室溫及光照下進行聚合反應。
反應體系中使用的惰性氣體為氬氣、氮氣和氦氣等,優選氬氣和氮氣。
在不同時間間隔時從反應器中取樣,樣品經過四氫呋喃、甲醇(或乙醚/正己烷)溶解/沉淀三次除雜,所得聚合物放入真空烘箱干燥至恒重。
上述技術方案中,所述光源為可見光,如led燈,氙燈和太陽光等。在實施中我們使用23w家用緊湊型節能燈兩只為光源,其中光源在420nm波長范圍處,光照強度為800μwcm-2。
上述技術方案中,所得聚合物的分子量分布窄(pdi=1.15~1.87),所得聚合物的實際相對分子量與理論分子量相符。
上述技術方案中,可以通過調整反應時間控制反應產物的分子量。
由于上述技術方案的應用,本發明與現有技術相比具有以下優點:
1、在室溫條件下,可以合成聚甲基丙烯酸甲酯、聚甲基丙烯酸芐酯、聚甲基丙烯酸羥丙酯和聚甲基丙烯酸n,n-二甲基氨基乙酯等聚合物,且聚合呈現“活性”/可控的特征。
2、本發明在光照下進行聚合反應,光化學過程可以通過光源的開關,快速地啟動或停止,并且特別適用于對熱和輻射敏感單體的聚合。
3、本發明使用苯甲醛衍生物類作為有機光氧化還原催化劑,具有無毒、廉價和易處理等優點。
4、本發明提供的室溫下光引發的苯甲醛衍生物/raft試劑催化的活性自由基聚合方法,工藝簡單、效率高和成本低。
5、本發明使用raft試劑作為鏈轉移劑,與催化劑結合,擴展了raft試劑與醛類催化劑調控聚合的單體范圍。
附圖說明
為了更清楚地說明本發明中的技術方案,下面將對方案中所需要使用的附圖做簡單的介紹。
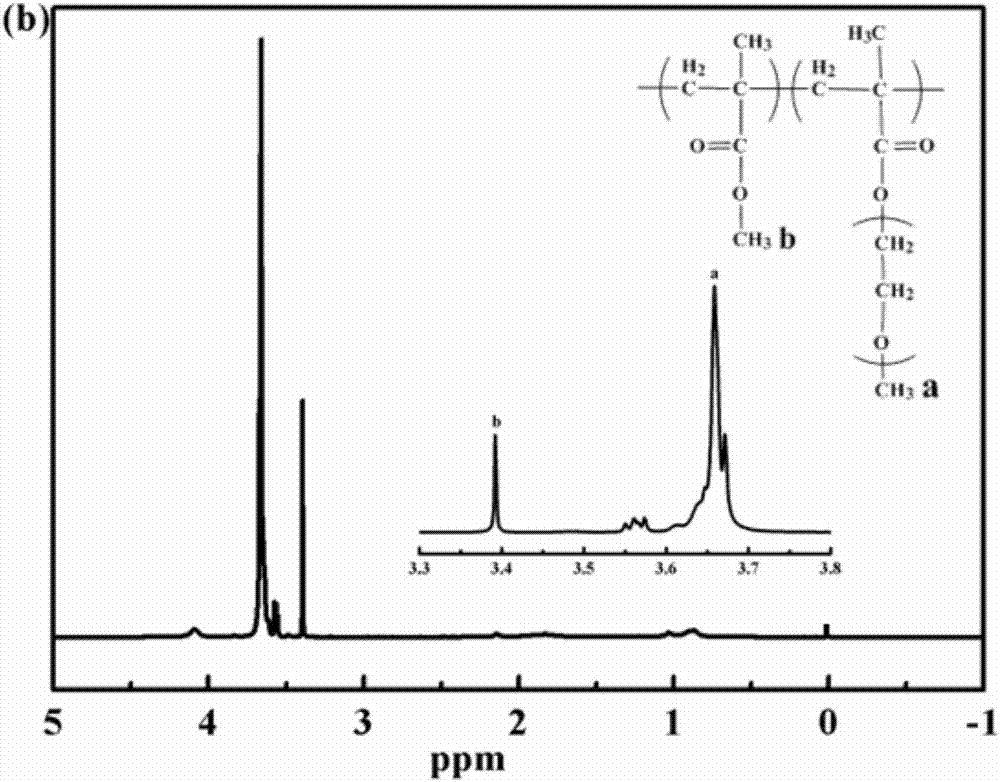
圖1a為實施例十一中大分子引發劑與嵌段共聚物的gpc曲線圖,圖1b為嵌段共聚物的核磁氫譜圖。
具體實施方式
下面將結合本發明實施例中的附圖,對本發明進行詳細的描述。實施例是為了更好地說明本發明的實施方法,并不限本發明的保護范圍。
實施例一:室溫條件下,2,4-二甲氧基苯甲醛為光氧化還原催化劑,4-氰基-4-(苯基硫代甲酰硫基)戊酸(ctp)為鏈轉移劑,dmf為溶劑,mma單體的光聚合。
按摩爾比[mma]0:[2,4-二甲氧基苯甲醛]0:[ctp]0=200:40:1,單體的質量分數為24wt%,依次加入2,4-二甲氧基苯甲醛(0.6639g)、ctp(0.0279g)、mma(2.00g)和dmf(6.00g)于25ml的圓底燒瓶中,加入攪拌子,經過3次冷凍-抽真空-充氬氣循環,在惰性氣氛下封瓶,之后將反應瓶安置于磁力攪拌器上,在室溫并在23w緊湊型節能燈(800μwcm-2@420nm)照射下開始反應。此后定時取樣并用四氫呋喃及甲醇(甲醇與水的體積比為7:3的混合液)溶解/沉淀三次,以除去樣品中未反應的單體及溶劑等,所得產物放入真空烘箱干燥至恒重即可得到聚甲基丙烯酸甲酯。計算產物的轉化率并通過gpc測定其分子量及分子量分布。
在該配比條件下,聚合物分子量隨轉化率的增長而線性增加。反應72h,單體轉化率達64.81%,聚合產物mn=15800gmol-1,pdi=1.41。
比較例一:
單體mma在n,n-二甲基甲酰胺(dmf)溶劑中的質量分數為24wt%。按照摩爾比[mma]0:[2,4-二甲氧基苯甲醛]0:[ctp]0=200/-/1以及200/-/-,分別將上述原料加入到25ml單支口圓底燒瓶中。其他操作參照實施例一。
當體系中不添加2,4-二甲氧基苯甲醛以及4-氰基-4-(苯基硫代甲酰硫基)戊酸時,反應96小時,幾乎無聚合物產生;當體系中不添加2,4-二甲氧基苯甲醛時,在鏈轉移劑4-氰基-4-(苯基硫代甲酰硫基)戊酸的存在下,有聚合物產生,但實驗結果表明,該反應只是普通的自由基聚合,分子量并不隨轉化率線性增長,即并非是可控自由基聚合。說明有機催化劑以及鏈轉移劑的存在對聚合反應的進行十分重要。
實施例二:室溫條件下,2,4-二甲氧基苯甲醛為光氧化還原催化劑,4-氰基-4-(苯基硫代甲酰硫基)戊酸(ctp)為鏈轉移劑,乙腈為溶劑時,mma單體的光聚合。
參照實施例一,按摩爾比[mma]0:[2,4-二甲氧基苯甲醛]0:[ctp]0=200:40:1,單體在溶劑中的質量分數為24wt%,依次加入樣品(底物用量與實施例一相同,只需改變溶劑)并采用與實施例一相同的處理方法處理產物,所得產物放入真空烘箱干燥至恒重即可得到聚甲基丙烯酸甲酯。反應166h,單體轉化率40.03%,聚合產物mn=6100gmol-1,pdi=1.46。
實施例三:室溫條件下,2,4-二甲氧基苯甲醛為光氧化還原催化劑,4-氰基-4-(苯基硫代甲酰硫基)戊酸(ctp)為鏈轉移劑,苯甲醚為溶劑時,mma單體的光聚合。
參照實施例一,按摩爾比[mma]0:[2,4-二甲氧基苯甲醛]0:[ctp]0=200:40:1,單體濃度為24wt%,依次加入樣品(底物用量與實施例一相同,只需改變溶劑)并采用與實施例一相同的處理方法處理產物,所得產物放入真空烘箱干燥至恒重即可得到聚甲基丙烯酸甲酯。反應98h,單體轉化率64.46%,聚合產物mn=11000gmol-1,pdi=1.66。
實施例四:室溫及光照條件下,對甲氧基苯甲醛為光氧化還原催化劑,4-氰基-4-(苯基硫代甲酰硫基)戊酸(ctp)為鏈轉移劑,mma單體的光聚合。
參照實施例一,按摩爾比[mma]0:[ctp]0=200:1,[mma]0:[對甲氧基苯甲醛]0=0.34:1,單體濃度為20wt%。不同之處在于,本實施例不加入溶劑,進行本體聚合。依次加入對甲氧基苯甲醛(8.00g)、ctp(0.0279g)和mma(2.00g)于25ml的圓底燒瓶中,并采用與實施例一相同的處理方法處理產物,所得產物放入真空烘箱干燥至恒重即可得到聚甲基丙烯酸甲酯。
在該配比條件下,聚合物分子量隨轉化率的增長而線性增加,且聚合物的分子量分布較窄(小于1.36),符合活性聚合的規律。反應125h,單體轉化率達85.34%,聚合產物mn=22600gmol-1,pdi=1.33。
比較例二:
單體mma在n,n-二甲基甲酰胺(dmf)溶劑中的質量分數為24wt%。按照摩爾比[mma]0:[對甲氧基苯甲醛]0:[ctp]0=200:40:1,依次加入對甲氧基苯甲醛(0.5446g)、ctp(0.0279g)、mma(2.00g)和dmf(6.00g)于25ml的圓底燒瓶中。其他操作參照實施例一。
在該配比條件下,聚合物分子量隨轉化率的增長而線性增加。反應97h,單體轉化率達50.17%,聚合產物mn=12500gmol-1,pdi=1.15。說明無論采用2,4-二甲氧基苯甲醛或對甲氧基苯甲醛,并且無論是溶液聚合或本體聚合,該體系都能引發甲基丙烯酸甲酯的活性自由基聚合。
實施例五:室溫及光照條件下,2,4-二甲氧基苯甲醛為光氧化還原催化劑,二硫代苯甲酸芐酯(bdb)為鏈轉移劑,dmf為溶劑,bnma單體的光聚合。
參照實施例一,按摩爾比[bnma]0:[2,4-二甲氧基苯甲醛]0:[bdb]0=200:40:1,單體濃度為24wt%,依次加入2,4二甲氧基苯甲醛(0.3773g)、bdb(0.0139g)、bnma(2.00g)和dmf(6.00g)于25ml的圓底燒瓶中并采用與實施例一相同的處理方法處理產物,所得產物放入真空烘箱干燥至恒重即可得到聚甲基丙烯酸芐酯。反應93h,單體轉化率76.29%,聚合產物mn=36800gmol-1,pdi=1.43。此外,在聚合過程中,聚合物分子量隨轉化率的增長而線性增加,并且聚合物的分子量分布較窄(小于1.44),符合活性聚合的規律。
實施例六:室溫及光照條件下,對甲氧基苯甲醛為光氧化還原催化劑,二硫代苯甲酸芐酯(bdb)為鏈轉移劑,bnma單體的光聚合。
參照實施例一,按摩爾比[bnma]0,:[bdb]0=200:1,[bnma]0:[對甲氧基苯甲醛]0=0.24:1,單體濃度為24wt%。同樣的,本實施例不加入溶劑,進行本體聚合。依次加入對甲氧基苯甲醛(6.3194g)、bdb(0.0139g)和bnma(2.00g)于25ml的圓底燒瓶中并采用與實施例一相同的處理方法處理產物,所得產物放入真空烘箱干燥至恒重即可得到聚甲基丙烯酸芐酯。
在該配比條件下,聚合物分子量隨轉化率的增長而線性增加,且聚合物的分子量分布較窄(小于1.28),符合活性聚合的規律。反應72h,單體轉化率達91.09%,聚合產物mn=31500gmol-1,pdi=1.24。
實施例七:室溫及光照條件下,對氰基苯甲醛為光氧化還原催化劑,二硫代苯甲酸異丙苯酯(cdb)為鏈轉移劑,dmf為溶劑,hpma單體的光聚合。
參照實施例一,按摩爾比[hpma]0:[對氰基苯甲醛]0:[cdb]0=200:50:1,單體濃度為24wt%,依次加入對氰基苯甲醛(0.4549g)、cdb(0.0189g)、hpma(2.00g)和dmf(6.00g)于25ml的圓底燒瓶中,其他操作參照實施例一。反應84小時,停止反應,樣品用四氫呋喃及乙醚溶解/沉淀三次,真空干燥至恒重,即可得到聚甲基丙烯酸羥丙酯。稱重法測得單體轉化率72.54%,聚合產物mn=13000gmol-1,pdi=1.21。此外,在聚合過程中,聚合物分子量隨轉化率的增長而線性增加,并且聚合物的分子量分布較窄(小于1.23),符合活性聚合的規律。
實施例八:室溫及光照條件下,對二甲氨基苯甲醛為光氧化還原催化劑,二硫代苯甲酸異丙苯酯(cdb)為鏈轉移劑,dmf為溶劑,hpma單體的光聚合。
參照實施例一,按摩爾比[hpma]0:[對二甲氨基苯甲醛]0:[cdb]0=200:50:1,單體濃度為23wt%,依次加入對二甲氨基苯甲醛(0.5174g)、cdb(0.0189g)、hpma(2.00g)和dmf(6.00g)于25ml的圓底燒瓶中,其他操作參照實施例一。反應125小時,停止反應,樣品用四氫呋喃及乙醚溶解/沉淀三次,真空干燥至恒重,即可得到聚甲基丙烯酸羥丙酯。稱重法測得單體轉化率65.23%,聚合產物mn=15600gmol-1,pdi=1.23。在聚合過程中,聚合物分子量隨轉化率的增長而線性增加,并且聚合物的分子量分布較窄(小于1.23),符合活性聚合的規律。
實施例九:室溫及光照條件下,2,4-二甲氧基苯甲醛為光氧化還原催化劑,s-正十二烷基-s'-(2-甲基-2-丙酸基)三硫代碳酸酯(ddmat)為鏈轉移劑,dmf為溶劑,dmaema單體的光聚合。
參照實施例一,按摩爾比[dmaema]0:[2,4-二甲氧基苯甲醛]0:[ddmat]0=150:50:1,單體濃度為23wt%,依次加入2,4-二甲氧基苯甲醛(0.7048g)、ddmat(0.0309g)、dmaema(2.00g)和dmf(6.00g)于25ml的圓底燒瓶中,其他操作參照實施例一。反應168小時,停止反應,樣品用四氫呋喃及正己烷溶解/沉淀三次,真空干燥至恒重,即可得到聚甲基丙烯酸n,n-二甲基氨基乙酯。稱重法測得單體轉化率86.54%,聚合產物mn=16100gmol-1,pdi=1.76。此外,在聚合過程中,聚合物分子量隨轉化率的增長而線性增加,并且聚合物的分子量分布較窄(小于1.76),符合活性聚合的規律。
實施例十:2,4-二甲氧基苯甲醛為光催化劑,mma單體的自身擴鏈反應
將mn=19000gmol-1,pdi=1.28的大分子引發劑macro-pmma(0.0140g),單體mma(1.00g),2,4-二甲氧基苯甲醛(0.3320g)以及thf(3.00g)依次加入干燥的單支口圓底燒瓶中,其他操作參照實施例一。反應72小時,停止反應,樣品用甲醇沉淀三次,真空干燥至恒重。稱重法測得單體轉化率為74.8%,gpc測得聚合物mn,=53900gmol-1,pdi=1.83。
實施例十一:2,4-二甲氧基苯甲醛為光催化劑,嵌段共聚物pmma-b-ppegma的合成
將mn=17000gmol-1,pdi=1.50的大分子引發劑macro-pmma(0.0358g),單體pegma(1.00g),2,4-二甲氧基苯甲醛(0.3495g)以及dmf(6.00g)依次加入干燥的單支口圓底燒瓶中,其他操作參照實施例一。反應132小時,停止反應,樣品用四氫呋喃和乙醚溶解/沉淀三次,真空干燥至恒重。稱重法測得單體轉化率為35.19%,gpc測得聚合物mn=64500gmol-1,pdi=1.87。嵌段共聚物pmma-b-ppegma的gpc曲線圖如圖1a所示,核磁氫譜如圖1b所示。
實施例十二:2,4-二甲氧基苯甲醛為光催化劑,嵌段共聚物pbnma-b-pmma的合成
將mn=34500gmol-1,pdi=1.28的大分子引發劑macro-pbnma(0.1725g),單體pmma(1.00g),2,4-二甲氧基苯甲醛(0.4716g)以及dmf(6.00g)依次加入干燥的單支口圓底燒瓶中,其他操作參照實施例一。反應體系在23w緊湊型節能燈照射下反應48小時,停止反應,樣品用甲醇沉淀三次,真空干燥至恒重。稱重法測得單體轉化率為42.13%,gpc測得聚合物mn,=52200gmol-1,pdi=1.65。
- 還沒有人留言評論。精彩留言會獲得點贊!