檢測靶核酸序列的引物、熒光探針及方法與流程
本發明屬于基因工程技術領域,更具體地,本發明涉及一種檢測靶核酸序列的引物、熒光探針及方法。
背景技術:
熒光pcr技術是檢測靶核酸序列的常用技術,目前常用的熒光pcr檢測方法包括taqman探針法、分子信標(molecularbeacon)探針法、熒光雜交雙探針(hybridizationprobe)法等。其中熒光雜交雙探針法主要通過使用兩條可與靶序列同一條單鏈雜交的熒光探針,其中一條探針3’端修飾一種熒光基團,另一條探針5’端修飾另一種熒光基團,當靶序列存在時通過非對稱pcr產生大量可與這兩條熒光探針反向互補的單鏈,當兩條熒光探針首尾相連與反向互補的單鏈dna雜交時,第一條探針3’端的熒光基團與第二條探針5’端的另一個熒光基團在空間距離上縮短,引發熒光能量共振轉移,所引起的熒光信號強弱變化可以用來檢測靶序列的存在。
熒光探針溶解曲線分析技術是基于不對稱pcr擴增技術的一種定性檢測方法。利用分子信標探針、fret探針或taqman探針在不對稱pcr反應后進行溶解曲線分析,可因為所結合序列變化所造成的溶解曲線峰值變化,從而分辨不同的靶序列位點。這項技術主要優勢在于可以利用不同熒光通道和不同熒光探針溶解峰溫度的組合同時檢測多個檢測位點,而且設計良好的探針可以通過溶解峰的變化分辨不同的突變位點。
目前熒光探針溶解曲線技術在同一熒光通道使用多條熒光探針進行檢測時,即使熒光探針未與靶序列結合,探針自身或探針之間在低溫條件下互相纏繞誤雜交也會產生背景信號,多條熒光探針的背景信號之間互相干擾疊加,造成熒光背景過高,從而影響了溶解曲線的判讀。
技術實現要素:
為了克服上述現有技術的缺陷,本發明提供了一種檢測靶核酸序列的引物、熒光探針及方法,該方法基于引物上淬滅基團對雜交探針熒光進行淬滅,使得多條檢測探針的背景信號之間不會互相干擾疊加。
為了實現上述發明目的,本發明采取了以下技術方案:
一種檢測靶核酸序列的引物,該引物的近3’端修飾有淬滅基團或熒光基團。
在其中一些實施例中,所述淬滅基團為bhq1、bhq2、或bhq3,所述熒光基團為fam、hex、rox、或cy5。
在其中一些實施例中,所述引物為正向引物或反向引物。
在其中一些實施例中,所述淬滅基團或熒光基團與所述引物的3’端末尾距離1-20個堿基。
在其中一些實施例中,所述淬滅基團或熒光基團與所述引物的3’端末尾距離1-10個堿基。
本發明還提供了一種檢測靶核酸序列的熒光探針,所述熒光探針與權利要求1~5任一項所述的引物與dna的不同單鏈互補,所述熒光探針的近3’端修飾有熒光基團或淬滅基團,且當所述引物修飾有熒光基團時,所述熒光探針修飾有淬滅基團,當所述引物修飾有淬滅基團時,所述熒光探針修飾有熒光基團。
在其中一些實施例中,所述淬滅基團為bhq1、bhq2、或bhq3,所述熒光基團為fam、hex、rox、或cy5。
在其中一些實施例中,所述熒光基團或淬滅基團與熒光探針的3’端末尾距離0-20個堿基。
在其中一些實施例中,所述熒光基團或淬滅基團與熒光探針的3’端末尾距離0個堿基,即直接修飾在3’端末尾。
本發明還提供了一種檢測靶核酸序列的方法,包括以下步驟:
以待檢測核酸序列為模板,以上述引物及熒光探針進行pcr反應,反應后進行溶解曲線分析,繪制溶解曲線,即可。
本發明的檢測靶核酸序列的熒光探針3’端修飾熒光基團,擴增靶序列的引物或通用引物3’端修飾淬滅基團。當檢測靶序列的pcr反應未進行時,熒光探針上的熒光基團處于正常狀態,可檢測到高強度的熒光信號。當檢測靶序列的pcr反應進行后,熒光探針與pcr反應過程產生的大量單鏈pcr產物結合,所發出的熒光被淬滅引物或通用淬滅引物上的淬滅基團淬滅,使熒光強度下降。當進行溶解曲線分析時,熒光探針在特定溫度下與所結合的單鏈pcr產物分離,所發出的熒光不再被淬滅引物或通用淬滅引物上的淬滅基團淬滅,從而熒光強度上升,形成特定溫度處的反向溶解峰。
當熒光基團修飾在引物或通用引物的3’端時,檢測靶核酸序列的探針3’端修飾淬滅基團。當檢測靶序列的pcr反應未進行時,引物上的熒光基團處于正常狀態,可檢測到高強度的熒光信號。當檢測靶序列的pcr反應進行后,修飾有淬滅基團的探針與pcr反應過程產生的大量單鏈pcr產物結合,引物上修飾的熒光基團發出的熒光被探針上的淬滅基團淬滅,使熒光強度下降。當進行溶解曲線分析時,探針在特定溫度下與所結合的單鏈pcr產物分離,引物上熒光基團所發出的熒光不再被探針的淬滅基團淬滅,從而熒光強度上升,形成特定溫度處的反向溶解峰。
與現有技術相比,本發明具有以下有益效果:
本發明的基于引物近3’端修飾的淬滅基團淬滅特異性熒光探針3’端熒光基團,或基于熒光探針近3’端修飾的淬滅基團淬滅特異性引物3’端熒光基團,并進行溶解曲線分析方法與其它溶解曲線分析技術相比,當檢測靶序列不存在情況下,熒光探針產生的熒光信號不受淬滅基團影響,熒光強度基本保持不變,使得雜交曲線分析時的背景本底明顯降低,不會影響其它陽性靶序列的檢測;當有多條熒光探針存在時,由于未結合的熒光探針在溫度變化時三維結構的改變不會像普通taqman探針或分子信標探針一樣明顯改變熒光信號強度,熒光探針之間的互相纏繞也不會像普通taqman探針或分子信標探針一樣互相改變熒光信號強度,使得探針之間的本底信號不會互相干擾,使得多重pcr的判讀更加清晰。
附圖說明
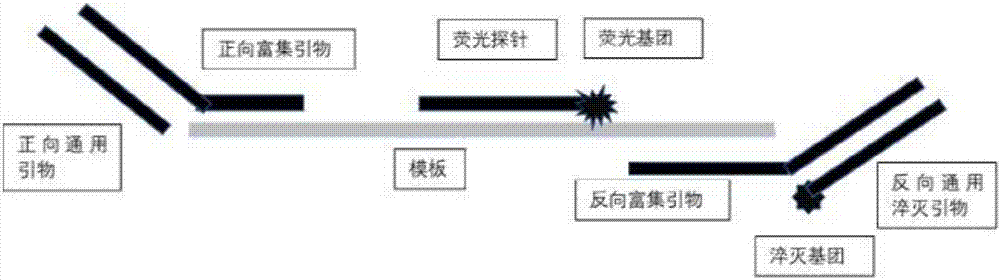
圖1為本發明實施例1的檢測靶核酸序列的引物及熒光探針的結構示意圖;
圖2為本發明實施例1的檢測靶核酸序列的通用引物及熒光探針的結構示意圖;
圖3為本發明實施例1的基于淬滅基團的引物和熒光探針雜交分析檢測方法的原理圖;
圖4為本發明實施例2的檢測結果圖;
圖5為本發明實施例2的檢測結果圖。
具體實施方式
下面結合附圖和具體實施例進一步敘述本發明,本發明未述及之處適用于現有技術。下面給出本發明的具體實施例,但實施例僅是為了進一步詳細敘述本發明,并不限制本發明的權利要求。
實施例1基于熒光能量共振轉移(fret)的檢測靶核酸序列的方法
請參閱圖1~2,為本發明的檢測靶核酸序列的引物或通用引物及熒光探針的結構示意圖。
引物可設計為正向引物或反向引物,本實施例中為反向引物,近3’端修飾有淬滅基團bhq1。熒光探針與引物與dna的不同單鏈互補,近3’端修飾有熒光基團fam,該熒光基團可被引物上的淬滅基團淬滅,使得熒光強度下降(圖1)。
正向和反向富集引物的3’端與模板反向互補,5’端為通用引物結合部位。通用引物可設計為正向引物或反向引物,本實施例中為反向,近3’端修飾有淬滅基團bhq2。熒光探針與通用引物對應的富集引物與dna的不同單鏈互補,近3’端修飾有熒光基團rox,該熒光基團可被通用引物上的淬滅基團上淬滅,使得熒光強度下降(圖2)。
請參閱圖3a~c,為本發明的基于淬滅基團的引物和熒光探針雜交分析檢測方法的原理圖;
在待檢測模板不存在情況下,熒光探針、通用引物均處于游離狀態,熒光基團與淬滅基團距離較遠,不發生fret效應,熒光基團所發出的熒光強度保持基本不變(圖3a)。
當待檢測模板存在的情況下,引物與待檢測模板結合,進行不對稱pcr反應,產生大量由引物延伸形成的單鏈pcr產物(圖3b)。
熒光探針與引物延伸形成的單鏈pcr產物互補結合,使得熒光基團與淬滅
基團相互靠近,發生fret現象,檢測到的熒光強度明顯下降。在進行溶解曲
線分析時,隨著溫度升高,熒光探針從結合的pcr產物上分離,fret現象消
失,熒光強度重新上升(圖3c)。
實施例2使用淬滅基團修飾的引物及熒光基團修飾的探針檢測不同濃度靶序列模板
根據本發明實施例1的原理,采用本實施例的方法檢測不同濃度靶序列模板,以評價其實用性。
靶序列模板為包含pcr擴增區域的人工質粒,由生工科技(上海)有限公司合成,按濃度稀釋為103/ul、104/ul不同濃度。
使用儀器為上海宏石slan-96p熒光定量pcr儀。
引物序列為:
std-f:tcagcaactagcttaccatc(seqidno:1),3’端倒數第2位t修飾bhq1淬滅基團
std-r:gctgacgttgcaagaagacg(seqidno:2)
熒光探針序列為:
std-p:tgatcatgtaatggaaggggcaaga(seqidno:3),3’端修飾hex熒光基團。
反應體系為:
反應條件:
熔解曲線分析:
設置檢測熒光通道為hex
反應條件為:95℃2min;45℃2min;45℃-85℃熔解曲線分析。
結果如圖3所示,從圖4可知,以103/ul、104/ul濃度質粒各1ul作為模板,反應后溶解曲線分析在63℃左右有明顯的倒置溶解峰,空白對照孔無對應的倒置溶解峰。
實施例3使用熒光基團修飾的引物及淬滅基團修飾的探針檢測不同濃度靶序列模板
根據本發明實施例1的原理,采用本實施例的方法檢測不同濃度靶序列模板,以評價其實用性。
靶序列模板為包含pcr擴增區域的人工質粒,由生工科技(上海)有限公司合成,按濃度稀釋為103/ul、104/ul不同濃度。
使用儀器為上海宏石slan-96p熒光定量pcr儀。
引物序列為:
std-f:tcagcaactagcttaccatc(seqidno:1),3’端倒數第2位t
修飾fam熒光基團
std-r:gctgacgttgcaagaagacg(seqidno:2)
熒光探針序列為:
std-p:tgtaatggaaggggcaaga(seqidno:3),3’端修飾bhq1淬滅基團。
反應體系為:
反應條件:
熔解曲線分析:
設置檢測熒光通道為fam
反應條件為:95℃2min;45℃2min;45℃-85℃熔解曲線分析。
結果如圖5所示,從圖5可知,以103/ul、104/ul濃度質粒各1ul作為模板,反應后溶解曲線分析在59℃左右有明顯的倒置溶解峰,空白對照孔無對應的倒置溶解峰。
試驗例1本發明的檢測方法的敏感性試驗
將淋球菌(ng)的質粒標準品分別做8個稀釋度從1×107~1×100拷貝數
/μl,以倍比稀釋為模板分別按實施例2的方法進行熒光定量pcr擴增,存在tm值63±1攝氏度的倒置溶解峰判讀為陽性,否則判讀為陰性。同時利用引物對seqidno:1和seqidno:2進行常規pcr擴增。
檢測結果表明,利用本發明的方法檢測ng標準質粒的靈敏度可達1×102拷貝數/μl,而普通pcr最低可以檢測到1×102拷貝數/μl,說明本發明的檢測方法與普通pcr的檢測方法相同。
試驗例2本發明的檢測方法的特異性試驗
通過本發明實施例2的檢測方法,分別對國家標準品菌種樣本:淋球菌、人乳頭瘤病毒、沙眼衣原體、淋球菌、微小脲原體、陰道毛滴蟲、生殖道支原體、人型支原體進行檢測,結果表明,除淋球菌標準品會有陽性溶解峰外,其他病原體樣本均沒有陽性溶解峰,證明本發明建立的檢測方法特異性強,與其他常見的生殖道病原體均無交叉反應。
試驗例3本發明的檢測方法的重復性試驗
以ng質粒標準品中的1×106和1×104拷貝數/μl兩個稀釋度的作為模板,對這兩個稀釋度在不同時間段進行2次重復測定,每次對同一模板同時進行3次重復測定及設置3個重復孔,按照本發明實施例2所述的方法進行實時熒光定量pcr。其中:變異系數(β)=標準偏差(sd)/平均數(x),結果重復性試驗陽性率100%。
以上所述實施例的各技術特征可以進行任意的組合,為使描述簡潔,未對上述實施例中的各個技術特征所有可能的組合都進行描述,然而,只要這些技術特征的組合不存在矛盾,都應當認為是本說明書記載的范圍。
以上所述實施例僅表達了本發明的幾種實施方式,其描述較為具體和詳細,但并不能因此而理解為對發明專利范圍的限制。應當指出的是,對于本領域的普通技術人員來說,在不脫離本發明構思的前提下,還可以做出若干變形和改進,這些都屬于本發明的保護范圍。因此,本發明專利的保護范圍應以所附權利要求為準。
序列表
<110>江西賢聚景欣醫藥生物科技有限公司
<120>檢測靶核酸序列的引物、熒光探針及方法
<130>3
<170>patentinversion3.1
<210>1
<211>20
<212>dna
<213>人工序列
<400>1
tcagcaactagcttaccatc20
<210>2
<211>20
<212>dna
<213>人工序列
<400>2
gctgacgttgcaagaagacg20
<210>3
<211>25
<212>dna
<213>人工序列
<400>3
tgatcatgtaatggaaggggcaaga25
- 還沒有人留言評論。精彩留言會獲得點贊!
- 用于檢測AEDs引起的SJS/TEN的引物、試劑盒及方法與制造工藝
- 檢測植原體的LAMP引物組及其試劑盒和應用的制造方法與工藝
- 用于多重PCR檢測致腹瀉寄生蟲的引物組及試劑盒的制造方法與工藝
- 使用重疊非等位基因特異性引物和等位基因特異性阻斷劑寡核苷酸的組合物進行等位基因特異性擴增的制造方法與工藝
- 一種基于特異性SS-COI引物的麗草蛉PCR快速檢測方法與制造工藝
- 一種利用線粒體D-loop區特異區域鑒定黿的標記引物及方法
- 一種用于檢測大豆擬莖點種腐病菌的特異性lamp引物組合物及其應用
- 一種針對膨脹污泥中Eikelboom Type 0092型絲狀菌群特異性引物和方法
- 紫苑石斛的分子特異性標記引物及檢測方法
- 一種檢測牛hcd攜帶者的特異性pcr引物、試劑盒及檢測方法
- 檢測植原體的LAMP引物組及其試劑盒和應用的制造方法與工藝
- 用于多重PCR檢測致腹瀉寄生蟲的引物組及試劑盒的制造方法與工藝
- 使用重疊非等位基因特異性引物和等位基因特異性阻斷劑寡核苷酸的組合物進行等位基因特異性擴增的制造方法與工藝
- 一種利用線粒體D-loop區特異區域鑒定黿的標記引物及方法
- 一種用于檢測大豆擬莖點種腐病菌的特異性lamp引物組合物及其應用
- 一種針對膨脹污泥中Eikelboom Type 0092型絲狀菌群特異性引物和方法
- 紫苑石斛的分子特異性標記引物及檢測方法
- 一種檢測牛hcd攜帶者的特異性pcr引物、試劑盒及檢測方法
- 用于檢測牛源性成分的lamp引物及其設計方法
- 七種不同種屬來源細胞的pcr檢測引物和檢測方法與應用
- 檢測植原體的LAMP引物組及其試劑盒和應用的制造方法與工藝
- 用于多重PCR檢測致腹瀉寄生蟲的引物組及試劑盒的制造方法與工藝
- 使用重疊非等位基因特異性引物和等位基因特異性阻斷劑寡核苷酸的組合物進行等位基因特異性擴增的制造方法與工藝
- 一種利用線粒體D-loop區特異區域鑒定黿的標記引物及方法
- 一種用于檢測大豆擬莖點種腐病菌的特異性lamp引物組合物及其應用
- 一種針對膨脹污泥中Eikelboom Type 0092型絲狀菌群特異性引物和方法
- 紫苑石斛的分子特異性標記引物及檢測方法
- 一種檢測牛hcd攜帶者的特異性pcr引物、試劑盒及檢測方法
- 用于檢測牛源性成分的lamp引物及其設計方法
- 七種不同種屬來源細胞的pcr檢測引物和檢測方法與應用