交聯多孔的La1?xCaxCoO3氧析出反應催化劑的制備方法與流程
本發明屬于堿性水電解催化劑研究領域,具體是一種交聯多孔的la1-xcaxcoo3氧析出反應催化劑的制備方法。
背景技術:
電解水時在陰極可生成氫氣,在陽極則析出氧氣,是可再生能源存貯與轉化的關鍵技術。堿性水電解過程中可使用非貴金屬氧化物作為陽極氧析出反應催化劑,常見的鈣鈦礦氧化物abo3有幾個關鍵特性:過渡金屬氧化態間的靈活變化形成氧化還原電對bm+/bm+1;氧空缺或氧過剩形成缺陷結構;優良的氧陰離子遷移率及氧交換動力學,均有利于催化氧還原/氧析出反應。abo3鈣鈦礦氧化物中a位離子的種類和比例的變化可改變晶體的結構、離子半徑/結合能及導電性;半徑較小的b位的過渡金屬離子可引起協同效應、形貌及引入混合價態等變化,調變了鈣鈦礦氧化物的電催化活性。另外,催化劑的比表面積越大催化活性越強,然而已有的鈣鈦礦氧化物催化劑在僅采用球磨、溶膠-凝膠或燃燒合成等方法制成時,比表面積低,粒徑較大,表面催化反應受到制約;有的鈣鈦礦氧化物催化劑還需要使用基底,添加物多,增加了成本。
技術實現要素:
本發明的目的是提供一種交聯多孔的la1-xcaxcoo3氧析出反應催化劑的制備方法,制備時無需使用模板和基底,制得的催化劑表面網狀多孔,比表面積大,催化活性高。
為實現上述目的,本發明采用的技術方案是:一種交聯多孔的la1-xcaxcoo3氧析出反應催化劑的制備方法,所述催化劑的通式為la1-xcaxcoo3,其中x為0.4或0.5,其制備方法包括以下步驟:
1)制液:稱取可溶性的la鹽、ca鹽及co鹽溶解于體積為v1的蒸餾水中,持續攪拌,制成金屬前驅體溶液a;稱取檸檬酸、造孔劑于體積為v2的蒸餾水中,制成溶液b;其中v1:v2=3:2;
2)混勻:在攪拌的條件下將溶液b逐漸加入金屬前驅體溶液a中形成混合溶液;
3)去除水分:將混合溶液恒溫蒸發去除水分,再在烘箱中烘干得到干凝膠;
4)制得催化劑:將干凝膠置于馬弗爐,在空氣氣氛下煅燒取出,得到黑色的交聯多孔的la1-xcaxcoo3催化劑。
具體地,所述步驟1)中可溶性的la鹽為la(no3)3.6h2o或lacl3,可溶性的ca鹽為ca(no3)2.4h2o或cacl2,可溶性的co鹽為co(no3)2.6h2o或cocl2.6h2o。
進一步地,所述步驟1)中可溶性的la鹽與可溶性的ca鹽的物質的量之和與可溶性的co鹽的物質的量相等,所述金屬前驅體溶液a的金屬離子的總濃度為0.5~1.0mol/l。
具體地,所述步驟1)中檸檬酸的物質的量與la鹽、ca鹽和co鹽的物質的量之和相等;造孔劑的物質的量是la鹽、ca鹽和co鹽的物質的量之和的1~2倍。
進一步地,所述步驟1)中造孔劑為蔗糖或葡萄糖。
進一步地,所述步驟3)中恒溫蒸發的溫度為80℃,時間為6~8h;烘干所用的溫度為100℃,時間為8~12h。
進一步地,所述步驟4)中煅燒的溫度為650~675℃,煅燒時間為1~3h。
進一步地,所述交聯多孔的la1-xcaxcoo3氧析出反應催化劑為蠕蟲狀不規則多孔形貌,催化劑的孔徑為6~100nm,催化劑的比表面積為18.6~44.8m2/g。
本發明1)本發明在制備過程中加入造孔劑,使制得的催化劑形成網狀多孔結構,增加催化劑的比表面積;造孔劑為蔗糖或葡萄糖,成本低;2)在煅燒過程中造孔劑碳化并在高溫燒除,無需加模板或基底,工藝流程簡單;3)制成的交聯多孔的la1-xcaxcoo3氧析出反應催化劑表觀活性可提高0.3~3.9倍,可應用于膜電極集合體。
附圖說明
圖1為本發明制備的交聯多孔la0.6ca0.4coo3催化劑的形貌圖。
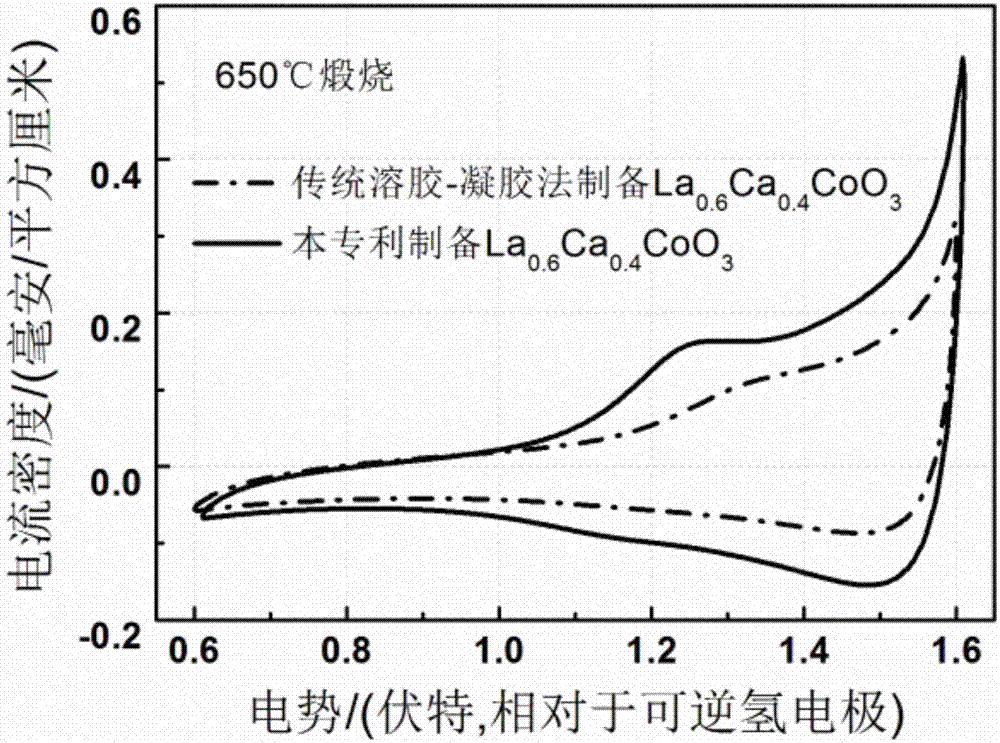
圖2為650℃煅燒條件下,本發明制備的交聯多孔la0.6ca0.4coo3催化劑與傳統溶膠-凝膠法制備的la0.6ca0.4coo3催化劑的循環伏安曲線對比圖。
圖3為650℃煅燒條件下,本發明制備的交聯多孔la0.6ca0.4coo3催化劑與傳統溶膠-凝膠法制備的la0.6ca0.4coo3催化劑的氧析出反應極化曲線對比圖。
圖4為675℃煅燒條件下,本發明制備的交聯多孔la0.6ca0.4coo3催化劑與傳統溶膠-凝膠法制備的la0.6ca0.4coo3催化劑的循環伏安曲線對比圖。
圖5為675℃煅燒條件下,本發明制備的交聯多孔la0.6ca0.4coo3催化劑與傳統溶膠-凝膠法制備的la0.6ca0.4coo3催化劑的氧析出反應極化曲線對比圖。
圖6為本發明制備的交聯多孔la0.5ca0.5coo3催化劑的形貌圖。
圖7為本發明制備的交聯多孔la0.5ca0.5coo3催化劑與傳統溶膠-凝膠法制備的la0.5ca0.5coo3催化劑的循環伏安曲線對比圖。
圖8為本發明制備的交聯多孔la0.5ca0.5coo3催化劑與傳統溶膠-凝膠法制備的la0.5ca0.5coo3催化劑的氧析出反應極化曲線對比圖。
圖9為傳統溶膠-凝膠法制備的la0.6ca0.4coo3催化劑的形貌圖。
具體實施方式
下面結合優選的具體實施方式對本發明作進一步詳細說明,便于更清楚地了解本發明,但本發明不局限于下述具體實施方式。
本發明在傳統的溶膠-凝膠法制備la1-xcaxcoo3催化劑的基礎上加入了蔗糖或葡萄糖作為造孔劑,在制備過程中不需使用模板,也不需要加入基底,在煅燒過程中造孔劑碳化并燒除,使制得的鈣鈦礦型氧化物催化劑la1-xcaxcoo3表面呈多孔結構,極大地增加了la1-xcaxcoo3的比表面積,應用于堿性水電解時電催化活性高。本發明中鈣鈦礦型氧化物催化劑la1-xcaxcoo3中所述元素組成1-x:x為經x射線熒光光譜分析(xrf)表征的實際la:ca原子比。
本發明的反應機理是:蔗糖在低溫條件(低于300℃)下首先碳化,隨后晶體以碳化生成的碳為模板,進一步在高溫下因碳的氧化脫除形成多孔結構;檸檬酸在反應中為絡合劑,能與金屬離子形成配合物,使離子分散均勻,不易團聚,從而形成金屬離子與檸檬酸根離子的網狀結構配合物;在高溫時,該絡合物開始進一步氧化緩慢釋放金屬離子以使摻雜金屬摻雜均勻,與此同時,金屬離子被氧化生成典型的鈣鈦礦型氧化物,該鈣鈦礦型氧化物中不含碳,是純凈的,且為典型的鈣鈦礦型單相結構。
本發明一種交聯多孔的la1-xcaxcoo3氧析出反應催化劑的制備方法,所述催化劑的化學式為la0.6ca0.4coo3或la0.5ca0.5coo3,其制備方法包括以下步驟:
1)制液:稱取可溶性的la(no3)3.6h2o或lacl3、ca(no3)2.4h2o或cacl2及co(no3)2.6h2o或cocl2.6h2o溶解于體積為v1的蒸餾水中,用磁力攪拌器持續攪拌,制成金屬前驅體溶液a,其中可溶性的la鹽與可溶性的ca鹽的物質的量之和與可溶性的co鹽的物質的量相等,金屬前驅體溶液a的金屬離子的總濃度為0.5~1.0mol/l;稱取檸檬酸、造孔劑蔗糖或葡萄糖于體積為v2的蒸餾水中,制成溶液b;其中v1:v2=3:2,檸檬酸的物質的量與la鹽、ca鹽和co鹽的物質的量之和相等;造孔劑的物質的量是la鹽、ca鹽和co鹽的物質的量之和的1~2倍;
2)混勻:在攪拌的條件下將溶液b逐滴依次加入金屬前驅體溶液a中形成混合溶液;
3)去除水分:將混合溶液于80℃恒溫蒸發去除大部分水分,再在烘箱中于100℃烘干得到干凝膠;
4)制得催化劑:將干凝膠置于馬弗爐,在空氣氣氛下煅燒取出,煅燒溫度為650~675℃,煅燒時間為1~3h,得到黑色的交聯多孔的la1-xcaxcoo3催化劑。
本發明的造孔劑可以為蔗糖或葡萄糖,兩者在催化劑的制備過程中的電化學效果類似,以下實施例優選蔗糖做造孔劑進行具體實施例的說明。
實施例1
1)交聯多孔鈣鈦礦型la0.6ca0.4coo3氧析出反應催化劑的制備
準確稱取0.6mmolla(no3)3.6h2o、0.4mmolca(no3)2.4h2o、1mmolco(no3)2.6h2o溶于30ml蒸餾水中,在磁力攪拌器上攪拌15min;另外稱取2mmol檸檬酸和3mmol蔗糖溶于20ml蒸餾水中并攪拌均勻形成溶液b;以1滴/秒的速度,在攪拌的條件下將溶液b逐滴加入金屬陽離子硝酸鹽混合溶液中形成溶膠溶液;將溶膠溶液置于磁力攪拌器中在80℃溫度下恒溫水浴攪拌,蒸去大部分水分形成濕凝膠;然后在烘箱內100℃烘干得到干凝膠;將干凝膠置于馬弗爐中,以空氣氣氛、650℃煅燒2h得到交聯多孔的la0.6ca0.4coo3催化劑粉末。
2)測試交聯多孔鈣鈦礦型la0.6ca0.4coo3氧析出反應催化劑的陽極性能與穩定性
采用三電極體系測試本發明方法所制備的la0.6ca0.4coo3催化劑。具體測試如下:分別以表面負載待測催化劑的玻碳電極為工作電極(催化劑擔載量為102μg/cm2),鉑片為對電極,hgo/hg電極作為參比電極,以0.1mol/l的氫氧化鉀溶液為電解液,將參比電極置于鹽橋中,使鹽橋一端的魯金毛細管尖端靠近工作電極,在氬氣飽和的電解液中以50mv/s掃描速度測試循環伏安曲線;在氧氣飽和的電解液中5mv/s掃描速度、電極轉速1600rpm測試催化劑對氧析出反應的催化活性;在o2飽和的電解液中,在恒定電勢1.6v(相對于可逆氫電極)、催化劑擔載量為127μg/cm2、電極轉速1600rpm及25℃條件下,通過氧析出時間-電流曲線測試催化劑穩定性。
對比例1
參照實施例1的催化劑的制備方法和測試方法,不同的是制液時不加入造孔劑,得到煅燒溫度為650℃的傳統溶膠-凝膠法制備的la0.6ca0.4coo3催化劑。
如圖1和圖9所示,使用場發射掃描電鏡(fe-sem)測試實施例1制得的la0.6ca0.4coo3催化劑和對比例1制得的la0.6ca0.4coo3催化劑的形貌相差較大,傳統溶膠-凝膠法制備的la0.6ca0.4coo3催化劑為不規則顆粒的集聚體,大小不一的微粒堆積形成一些孔洞;實施例1中制得的la0.6ca0.4coo3催化劑表面則致密排布蠕蟲狀孔洞,孔壁相互交聯。bet法測試本實施例1所得催化劑的表面積為18.6m2/g,是傳統溶膠-凝膠法制備la0.6ca0.4coo3催化劑bet表面積(11.1m2/g)的1.6倍,比表面積明顯增大。
實施例1和對比例1測試陽極性能與穩定性所得結果如下表1所示:
表1
如圖2所示,實施例1和對比例1制得的la0.6ca0.4coo3催化劑測試所得的循環伏安曲線表明:實施例1制得的la0.6ca0.4coo3催化劑的循環伏安電量比對比例1制得的la0.6ca0.4coo3催化劑明顯增大,說明加入造孔劑蔗糖可將la0.6ca0.4coo3催化劑的電化學活性面積提高約0.6倍。
如圖3所示,實施例1和對比例1制得的催化劑的氧析出反應極化曲線對比圖表明:在1.7v電勢下,實施例1制得的la0.6ca0.4coo3催化劑的電流密度是對比例1制得的la0.6ca0.4coo3催化劑的2.1倍,說明加入造孔劑蔗糖使la0.6ca0.4coo3催化劑的表觀活性明顯提高。
實施例2
參照實施例1的催化劑的制備方法和測試方法,不同的是煅燒溫度為675℃,得到交聯多孔的la0.6ca0.4coo3催化劑粉末。
對比例2
參照實施例2的催化劑的制備方法和測試方法,不同的是制液時不加入造孔劑,得到煅燒溫度為675℃的傳統溶膠-凝膠法制備的la0.6ca0.4coo3催化劑。
測試實施例2和對比例2的bet表面積,實施例2的bet表面積為44.8m2/g,是傳統溶膠-凝膠法制備la0.6ca0.4coo3催化劑bet表面積(12.2m2/g)的約3.7倍。
實施例2和對比例2測試陽極性能與穩定性所得結果如下表2所示:
表2
如圖4所示,實施例2和對比例2制得的la0.6ca0.4coo3催化劑測試所得的循環伏安曲線表明:實施例2制得的la0.6ca0.4coo3催化劑的循環伏安電量是對比例2制得的la0.6ca0.4coo3催化劑的4.7倍,說明加入造孔劑蔗糖可將la0.6ca0.4coo3催化劑的電化學活性面積顯著提高。
如圖5所示,實施例2和對比例2制得的催化劑的氧析出反應極化曲線對比圖表明:在1.7v電勢下,實施例2制得的la0.6ca0.4coo3催化劑的電流密度是對比例2制得的la0.6ca0.4coo3催化劑的4.9倍,說明加入造孔劑蔗糖使la0.6ca0.4coo3催化劑的表觀活性明顯提高。
實施例3
參照實施例2的制備方法和測試方法,不同的是在制液過程中加入造孔劑蔗糖的量為2mmol,與檸檬酸一起溶于30ml蒸餾水中;最終制得交聯多孔的la0.6ca0.4coo3催化劑粉末。
實施例4
參照實施例2的制備方法和測試方法,不同的是在制液過程中加入造孔劑蔗糖的量為3mmol,與檸檬酸一起溶于30ml蒸餾水中;最終制得交聯多孔的la0.6ca0.4coo3催化劑粉末。
實施例5
參照實施例2的制備方法和測試方法,不同的是在制液過程中加入造孔劑蔗糖的量為4mmol,與檸檬酸一起溶于30ml蒸餾水中;最終制得交聯多孔的la0.6ca0.4coo3催化劑粉末。
實施例3~5制得的交聯多孔的la0.6ca0.4coo3催化劑和對比例2的la0.6ca0.4coo3催化劑對應測得的循環伏安電量、氧析出催化活性比較見下表3。
表3
由表3可知,與傳統溶膠-凝膠法制備la0.6ca0.4coo3催化劑相比,利用造孔劑制備的交聯多孔la0.6ca0.4coo3催化劑的電化學活性面積明顯提高,其表觀活性成比例增加。
實施例6
參照實施例2的制備方法和測試方法,不同的是在金屬前驅體溶液a的制液過程中稱取0.5mmolla(no3)3.6h2o、0.5mmolca(no3)2.4h2o、1mmolco(no3)2.6h2o溶于30ml蒸餾水中;最終制得交聯多孔的la0.5ca0.5coo3催化劑粉末。
對比例3
參照實施例6的催化劑的制備方法和測試方法,不同的是制液時不加入造孔劑,得到煅燒溫度為675℃的傳統溶膠-凝膠法制備的la0.5ca0.5coo3催化劑。
如圖6所示,使用場發射掃描電鏡(fe-sem)測試實施例6制得的la0.5ca0.5coo3催化劑的形貌,實施例6中制得的la0.5ca0.5coo3催化劑致密排布蠕蟲狀孔洞,孔壁相互交聯。bet法測試本實施例6所得催化劑的表面積為28.0m2/g,是傳統溶膠-凝膠法制備la0.5ca0.5coo3催化劑bet表面積(11.6m2/g)的2.4倍。
如圖7所示,實施例6和對比例3制得的la0.5ca0.5coo3催化劑測試所得的循環伏安曲線表明:實施例6制得的la0.5ca0.5coo3催化劑的循環伏安電量為0.85mc/cm2,是對比例3制得的la0.5ca0.5coo3催化劑(0.45mc/cm2)的1.9倍,說明加入造孔劑蔗糖可將la0.5ca0.5coo3催化劑的電化學活性面積提高。
如圖8所示,實施例6和對比例3制得的催化劑的氧析出反應極化曲線對比圖表明了:在1.7v電勢下,實施例6制得的la0.5ca0.5coo3催化劑的電流密度(1.98ma/cm2)是對比例3制得的la0.5ca0.5coo3催化劑(1.02ma/cm2))的1.9倍,說明加入造孔劑蔗糖使la0.5ca0.5coo3催化劑的表觀活性明顯提高。
本說明書中未詳細說明的內容為本領域普通技術人員公知的現有技術。
以上所述的具體實施方式僅僅是示意性的,本發明中所用到的技術術語的限定性修飾詞僅為方便本發明的描述,本領域的普通技術人員在本發明交聯多孔的la1-xcaxcoo3氧析出反應催化劑的制備方法的啟示下,在不脫離本發明宗旨和權利要求所保護的范圍情況下,還可衍生出很多形式,這些均在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!