一種毛細管動態涂層的制備方法與流程
本發明涉及一種毛細管動態涂層的制備方法。
背景技術:
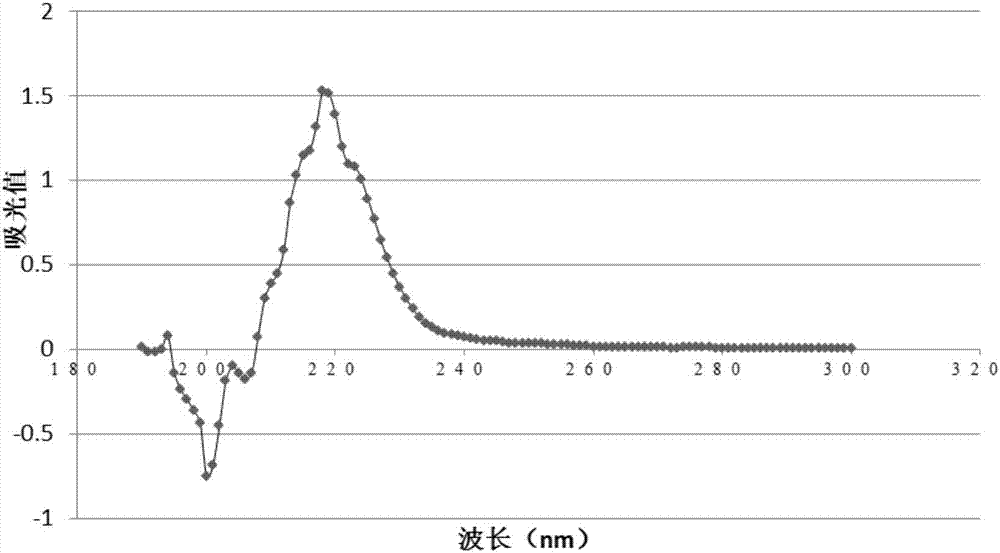
:毛細管電泳儀作為一種分離分析儀器,具備高效、快速、進樣少、污染少、自動化程度高等特點,廣泛應用于氨基酸、多肽、蛋白質、核酸等物質的分離。但是由于毛細管儀在電泳過程中產生的電滲流不穩定,另外,內壁的負電性和親水性導致分析物極易被吸附,極大地影響蛋白質和多肽分離的重現性和分離效率。一般認為,蛋白質等電點在8以上或者分子量超過50kda即難以在裸露的毛細管中進行分析。為了克服毛細管壁的吸附作用,許多方法已被采用,如使用極端ph值緩沖液、添加高離子強度的電解質以及涂層技術等。前二者會限制分離的選擇性并可能導致蛋白變性。涂層技術指在毛細管內壁以物理或化學方法形成不同性質的涂層。毛細管涂層的形成方法大致可以分為三類,即動態吸附、物理涂布和化學鍵合。化學和物理涂層法主要通過物理吸附或化學交聯等方法在管壁內側形成單分子層或交聯的涂層,掩蔽毛細管內壁的負電荷,從而達到抑制吸附的目的。涂層毛細管會隨使用次數的增加而受損,對應的分離重現性也相應變差。動態涂層指將涂層材料添加到緩沖液中,不斷吸附到毛細管內壁,以補充管壁的解吸附材料,維持涂層穩定。因此,動態涂層具有較好的重現性和分離效果。目前文獻報道的陽離子多聚物涂層材料大多需要實驗室自行合成,涂層制作過程相對復雜,涂層材料耐受酸堿的能力有限,并未找到一種對所有種類蛋白質的分離都有效的普適性涂層。因此,尋求新的涂層材料,簡化涂層制作技術,不斷提高其分離復雜蛋白成分的能力,是毛細管動態涂層研究的一個重要方向。技術實現要素:本發明的一個目的是提供一種毛細管動態涂層的制備方法。本發明所提供的毛細管動態涂層的制備方法,包括下述步驟:1)對毛細管進行活化;2)用陽離子型聚乙烯吡咯烷酮水溶液沖洗活化后的毛細管,然后運行緩沖液進行沖洗。其中,所述陽離子型聚乙烯吡咯烷酮(cpvp)水溶液的質量-體積濃度為0.001-0.5%(g/100ml)。具體的cpvp濃度可根據使用情況進行調節。當采用cpvp動態涂層檢測電滲流時,因為很低濃度cpvp即可調節電滲流,所以cpvp水溶液的質量-體積濃度選擇0.001-0.25%(g/100ml)。當采用cpvp動態涂層分離蛋白時,由于抑制毛細管壁吸附需要更高濃度的cpvp,所以cpvp水溶液的質量-體積濃度選擇0.1-0.5%(g/100ml)。所述緩沖液為磷酸鹽緩沖液,所述磷酸鹽緩沖液的濃度可為10-50mm,具體可為10mm、20mm、30mm、40mm或50mm;ph值可為3.0-8.0,具體可為ph3.0、ph6.5、ph7.0、ph7.5或ph8.0。所述用陽離子型聚乙烯吡咯烷酮水溶液沖洗毛細管的沖洗壓力可為15-25psi,優選20psi,沖洗時間為10-20分鐘。所述緩沖溶液的沖洗壓力為可為15-25psi,優選20psi;沖洗時間為10-20分鐘。為了使涂層更好的平衡,所述步驟2)中,用陽離子型聚乙烯吡咯烷酮的水溶液沖洗毛細管后運行緩沖液之前,還包括靜置的步驟;所述靜置的時間為4-10分鐘。利用上述具有動態涂層的毛細管分離蛋白時,為了減少蛋白對毛細管壁的吸附,所述方法還包括:對所制備的動態涂層用高電壓進行預分離的步驟;所述高電壓為10-12kv,分離的時間為2-4分鐘。本發明中所述陽離子型聚乙烯吡咯烷酮是由二甲基二烯丙基氯化銨和乙烯吡咯烷酮共聚得到的。所述陽離子化的聚乙烯吡咯烷酮的粘均分子量可為20000-25000。所述步驟1)對毛細管進行活化的方法可按照本領域常規的活化方法進行,具體步驟可參考下述方法:1mnaoh20psi沖洗15-20分鐘,超純水20psi沖洗10分鐘,運行緩沖液20psi沖洗20-30分鐘備用。所述運行緩沖液為磷酸鹽緩沖液,所述磷酸鹽緩沖液的濃度可為10-50mm,ph值可為3.0-8.0。所述陽離子型聚乙烯吡咯烷酮具體可按照如下方法制備得到:在裝有磁力攪拌器、溫度計和通n氣管的反應器中加入一定量的二甲基二烯丙基氯化銨(diallyldimethylammoniumchloriddadmac)、乙烯吡咯烷酮(vinylpyrrolidonvp)(二者質量比為1:4)、引發劑2,2一偶氮二異丁基脒二鹽酸鹽(為單體質量的0.25%),加入一定量蒸餾水溶解,使vp濃度為0.5g/ml。升溫至65℃,反應5h,得到膠體,加適量水溶解,然后用丙酮沉淀。濾出沉淀并真空干燥即為目標共聚物。上述方法制備得到的具有動態涂層的毛細管也屬于本發明的保護范圍。本發明的再一個目的是提供上述具有動態涂層的毛細管在毛細管電泳分離蛋白質中的應用。所述蛋白質優選為堿性蛋白質。所述蛋白質具體可選自下述至少兩種:牛胰凝乳蛋白酶原、核糖核酸酶a、雞心細胞色素c、肌紅蛋白和伴清蛋白。所述毛細管電泳分離上述蛋白質的電泳條件為:分離電壓反向12kv,檢測波長214nm,毛細管溫度25℃,樣品托盤溫度10℃,電泳時間45-50分鐘。由于聚乙烯吡咯烷酮極易被水沖掉,不適合用于動態涂層,一般只能作為毛細管共價涂層材料,限制了其使用范圍。而本發明采用陽離子化的聚乙烯吡咯烷酮(cationicpolyvinylpyrrolidonecpvp)作為涂層材料,對毛細管柱進行動態修飾,這種陽離子化的涂層材料能夠穩定反向電滲流。同時通過對毛細管電泳運行中的各項參數進行優化,建立了一個穩定涂層制備的實驗流程和技術方法,可控制電滲流、抑制管壁吸附,有效地提升毛細管電泳分離堿性蛋白的能力,分離效率最高達3.2×105。附圖說明圖1為實施例1制備的cpvp的紫外吸收光譜圖。圖2為cpvp濃度對電滲流的影響圖。圖3為ph值對電滲流的影響圖。圖4為50mmph6.5磷酸鹽緩沖液電泳分離蛋白標準品混合物;其中1:伴清蛋白;2:肌紅蛋白;3:牛胰凝乳蛋白酶原;4:核糖核酸酶a;5:雞心細胞色素c。圖5為不同濃度運行緩沖液電泳分離蛋白標準品混合物。具體實施方式下述實施例中所使用的實驗方法如無特殊說明,均為常規方法。下述實施例中所用的材料、試劑等,如無特殊說明,均可從商業途徑得到。實施例1、cpvp的制備及性能測試1.cpvp的制備依據文獻報道(林媚林美玉朱煒等,陽離子型聚乙烯吡咯烷酮的制備及其負載rna性能。應用化學,2014,第31卷第6期。),合成該化合物。具體方法為:在裝有磁力攪拌器、溫度計和通n氣管的反應器中加入一定量的二甲基二烯丙基氯化銨(diallyldimethylammoniumchloriddadmac)、乙烯吡咯烷酮(vinylpyrrolidonvp)(二者質量比為1:4)、引發劑2,2一偶氮二異丁基脒二鹽酸鹽(為單體質量的0.25%),加入一定量蒸餾水溶解,使vp濃度為0.5g/ml。升溫至65℃,反應5h,得到膠體,加適量水溶解,然后用丙酮沉淀。濾出沉淀并真空干燥即為目標共聚物。2.cpvp的粘均測試先配置1m的氯化鈉溶液500ml以上,以后所有溶劑均以此為溶劑計算。具體:稱取58.5g氯化鈉,加入水約970ml,最后定容為1000ml;用鹽水溶液配置c1:0.01g/ml的聚合物溶液50ml(確定濃度)和c2:0.04g/ml。具體:稱取cpvp0.51215g,用1m氯化鈉溶解過夜,最后定容為50ml,得到濃度為c1=0.01024g/ml;稱取cpvp2.01601g,用1m氯化鈉溶解過夜,最后定容為50ml,得到濃度為c2=0.04032g/ml;在30℃恒溫浴槽內測試,用烏式粘度計測量純氯化鈉溶液的流出時間記為t0:測量聚合物溶液c1的流出時間為t5。將c2溶液稀釋配制出0.002;0.004;0.006;0.008g/ml的溶液各50ml。清洗粘度管,最后用少量被測試溶液潤洗粘度管3次后再加入被測試液,測試溶液的流出時間,并對應濃度與時間。具體:量取c1溶液10ml,用鹽水定容為50ml;量取c1溶液20ml,用鹽水定容為50ml;量取c2溶液7.5ml,用鹽水定容為50ml;量取c2溶液10ml,用鹽水定容為50ml;因此各樣品濃度分別為1號2.048mg/ml;2號為4.096mg/ml;3號為6.048mg/ml;4號為8.064mg/ml;cpvp的粘均分子量烏式粘度計測得不同樣品濃度的比粘度、相對對數粘度分別為:外推到0濃度時的比濃增比粘度為0.01592毫升/毫克=0.01592升/克=0.1592分升/克,根據mark—houwink公式[η]=kmα,即得到粘均相對分子質量。計算cpvp相對分子質量時k=1.4x10-4,α=0.7來計算,可以得到其粘均分子量為:(0.1592/1.4x10-4)^(1/0.7)=2.3x104,因此該聚合物的相對粘均分子量約為23000。3.cpvp的紫外光譜特性測定取質量體積分數為0.5%cpvp水溶液,利用紫外分光光度計測定其在200-300nm的光吸收,獲得該物質的紫外吸收光譜。其紫外吸收光譜圖如圖1所示。從圖1可以看出,cpvp在紫外區有光吸收,最大吸收波長位于218nm,因此,在電泳過程中不宜將cpvp添加到運行緩沖液中。實施例2、制備毛細管cpvp動態涂層系統1.1蛋白標準品的制備牛胰凝乳蛋白酶原(pi9.3,分子量25000da),核糖核酸酶a(pi7.8,分子量13700da),雞心細胞色素c(pi10.8,分子量12400da),肌紅蛋白(pi7.3,分子量16700da),伴清蛋白(pi6.8,分子量77770da),均購自sigmachemicals。將5種蛋白標準品分別用去離子水配制成10mg/ml溶液,-80度長期保存,4度保存一周,用時等量混合終濃度為1mg/ml。1.2cpvp動態涂層系統分離蛋白標準品運行方法河北永年石英毛細管(40.2cm,有效長度30cm,毛細管內徑50μm)。新管活化:1mnaoh20psi沖洗20分鐘,超純水20psi沖洗10分鐘,運行緩沖液20psi沖洗20分鐘備用。所述緩沖液為不同濃度(10-50mm)和ph值(ph=3.0,ph=6.5,ph=7.0,ph=7.5,ph=8.0)的磷酸鹽緩沖液。動態涂層制備質量體積分數為0.5%的cpvp水溶液20psi沖洗10分鐘,靜置4分鐘,運行緩沖液20psi沖洗10分鐘,高電壓12kv預分離2分鐘。所述緩沖液為不同濃度(10-50mm)和ph值(ph=3.0,ph=6.5,ph=7.0,ph=7.5,ph=8.0)的磷酸鹽緩沖液。進樣樣品濃度:1mg/ml,壓力進樣0.5psi,3秒。毛細管電泳分離電壓反向12kv,檢測波長214nm,毛細管溫度25℃,樣品托盤溫度10℃,電泳時間45分鐘。1.3動態涂層檢測電滲流的運行方法河北永年石英毛細管(40.2cm,有效長度30cm,毛細管內徑50μm)。新管活化:1nnaoh20psi沖洗20分鐘,超純水20psi沖洗10分鐘,運行緩沖液20psi沖洗20分鐘備用。所述緩沖液為不同濃度(10-50mm)和ph值(ph=3.0,ph=6.5,ph=7.0,ph=7.5,ph=8.0)的磷酸鹽緩沖液。動態涂層制備不同濃度(0.001-0.25%)的cpvp水溶液20psi沖洗5分鐘,運行緩沖液(ph=6.5,20mm磷酸鹽緩沖液)20psi沖洗3分鐘。進樣樣品濃度:0.1%(w/v)二甲基亞砜(dmso),壓力進樣0.5psi,5秒。毛細管電泳分離電壓反向20kv,檢測波長214nm,毛細管溫度25℃,樣品托盤溫度10℃,電泳時間10分鐘。1.4數據分析利用pa800plus自帶的32karat軟件計算理論塔板數。遷移時間相對標準偏差計算,根據公式:相對標準偏差(rsd)=標準偏差(sd)/計算結果的算術平均值(x)*100%2結果與討論2.1cpvp濃度對電滲流的影響以ph=6.5,20mm磷酸鹽緩沖液為運行緩沖液,分別檢測了0.001-0.25%cpvp動態涂層濃度對電滲流的影響,結果如圖2所示,cpvp的濃度為0.001-0.1%時,電滲流逐漸增大,當cpvp的濃度大于0.1%時,電滲流開始下降。2.2ph值對電滲流的影響由于磷酸鹽的緩沖范圍分別在ph(1.12-3.12)、(6.20-8.20),故分別選取ph3.0、ph6.5、ph7.0、ph7.5、ph8.0時的50mm磷酸鹽緩沖液為運行緩沖液,以0.1%dmso水溶液為中性標記物,檢測了不同ph值磷酸鹽緩沖液為介質,cpvp動態涂層的電滲流。結果如圖3所示,隨著ph值增加,電滲流逐漸減少。2.3運行緩沖液濃度對cpvp動態涂層系統分離柱效的影響分別以不同濃度(10-50mm)ph6.5磷酸鹽緩沖液為運行緩沖液,分離蛋白標準品混合物,結果見圖4-5,并用32karat軟件計算理論塔板數(n)定量各種蛋白的分離柱效。結果如表1所示,對于不同蛋白,其最適運行緩沖液濃度略有所不同。如牛胰凝乳蛋白酶原和核糖核酸酶a在50mm的ph6.5磷酸鹽緩沖液分離柱效最高,分別可達2.9x105和1.7x105;而伴清蛋白、肌紅蛋白、雞心細胞色素c在各個濃度下分離柱效差別不大,基本維持在同樣數量級。表1:運行緩沖液濃度對cpvp動態涂層系統分離柱效的影響n=32.4ph值對cpvp動態涂層系統分離柱效的影響分別選取ph3.0、ph6.5、ph7.0、ph7.5、ph8.0時的50mm磷酸鹽緩沖液為運行緩沖液,電泳分離蛋白標準品混合物,并用32karat軟件計算理論塔板數定量各種蛋白的分離柱效。結果如表2所示,在這些ph環境下,各種蛋白的分離柱效略有變化,其中牛胰凝乳蛋白酶原在ph8.0和ph6.5時,分離柱效可達105,而在其余ph環境中,分離柱效僅達104。表2:ph值對cpvp動態涂層系統分離柱效的影響n=32.5cpvp動態涂層系統的重現性和穩定性為了評價cpvp動態涂層系統的重現性和穩定性,分別檢測重復進樣時電滲流和蛋白遷移時間的變化。結果顯示在不同濃度、不同ph值情況下,重復進樣6次電滲流中性標記物dmso遷移時間的重現性rsd<3%(表3a)。對于蛋白標準品連續進樣6次遷移時間的重現性rsd<3%,每日更換毛細管,連續進樣4天,每天進樣4-6次,蛋白標準品遷移時間的重現性rsd<6%(表3b),說明該動態涂層系統具有很好的重現性和穩定性。表3a:電滲流中性標記物遷移時間的重現性和穩定性cpvp濃度0.001%0.005%0.01%0.05%0.1%0.25%rsd(%)n=60.080.090.050.060.080.09ph值3.06.57.07.58.0rsd(%)n=61.050.110.480.202.67表3b:蛋白遷移時間的重現性和穩定性在本說明書的描述中,參考術語“一個實施例”、“一些實施例”、“示例”、“具體示例”、或“一些示例”等的描述意指結合該實施例或示例描述的具體特征、結構、材料或者特點包含于本發明的至少一個實施例或示例中。在本說明書中,對上述術語的示意性表述不必須針對的是相同的實施例或示例。而且,描述的具體特征、結構、材料或者特點可以在任一個或多個實施例或示例中以合適的方式結合。此外,在不相互矛盾的情況下,本領域的技術人員可以將本說明書中描述的不同實施例或示例以及不同實施例或示例的特征進行結合和組合。盡管上面已經示出和描述了本發明的實施例,可以理解的是,上述實施例是示例性的,不能理解為對本發明的限制,本領域的普通技術人員在本發明的范圍內可以對上述實施例進行變化、修改、替換和變型。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1