作為具有形態發生活性的包被層和支架的非晶形無機聚磷酸鹽?磷酸鈣和碳酸鈣顆粒的制作方法
本發明涉及制備基于無機聚磷酸鹽(聚p)和磷酸鈣或碳酸鈣的顯示成骨活性的非晶形、納米顆粒材料或微粒材料的方法。在本發明的一個方面,本發明人顯示非晶形聚磷酸鈣(ca-聚p)微粒可用于鈦合金表面的生物功能化。本發明的方法允許在鈦氧化的ti-6al-4v支架上制造耐久且穩定的、幾乎均勻且具有形態發生活性的ca-聚p層,與生物惰性的非改性鈦表面相比,其支持骨細胞的生長并增強骨細胞的功能活性。優選的方面涉及在低濃度聚磷酸鈉(na-聚p)的存在下形成非晶形磷酸鈣(cap)顆粒。這種材料導致參與骨形成的蛋白的表達強烈上調。本發明的另一方面涉及含有聚p穩定的acc和少量球霰石的材料,該材料在動物實驗中顯示成骨活性并支持植入區域的再生。本發明的非晶形材料具有用于骨植入物的潛力。
背景技術:
除了膠原蛋白和水之外,骨骼的基本構成塊還包含碳酸化磷灰石[ca5(po4,co3)3(oh)]以及羥基磷灰石(ha)。結晶礦物質可能由非晶形磷酸鈣(acp)形成(wangy,etal.(2013)water-mediatedstructuringofboneapatite.natmater12:1144-1153)。
最近的證據表明非晶形碳酸鈣(acc)作為用于形成acp和碳酸化磷灰石的生物種子,其是由碳酸酐酶(ca),很可能是由可溶性ca-ii同種型和/或細胞膜相關ca-ix形成的材料(wangxh,etal.(2014)modulationoftheinitialmineralizationprocessofsaos-2cellsbycarbonicanhydraseactivatorsandpolyphoshate.calciftissueint94:495-509;müllerweg,etal.mineralizationofbone-relatedsaos-2cellsunderphysiologicalhypoxicconditions.febsj.2015oct10)。
acc是碳酸鈣最不穩定的多晶型物,其存在于非晶形相和結晶相中;在三種主要的結晶多晶型物球霰石、霰石和方解石中,亞穩態球霰石是結晶caco3的熱力學最不穩定形式(meldrumfc,
因此,可以將骨ha的形成細分為以下三個機械上不同的階段:
1.通過碳酸酐酶來酶促形成acc生物種子;
2.在acp形成下,通過磷酸鹽來非酶促交換碳酸鹽離子;和
3.將acp轉化為結晶相碳酸化磷灰石/ha。
acc作為潛在的誘導再生/支撐材料的應用,受到acc本身不穩定這一事實的阻礙。acc在體內的穩定性通過通常與mg2+結合的專門蛋白調節,而在體外條件下,非生物來源添加劑如可溶性聚羧酸鹽,還有mg2+、三磷酸鹽或聚磷酸鹽種類,將acc凍結到相對穩定的相(kellermeierm,etal.(2010)stabilizationofamorphouscalciumcarbonateininorganicsilica-richenvironments.jamchemsoc132:17859-17866)。
相反,球霰石足夠穩定以允許解離,反過來又可以作為骨再生的潛在離子緩沖系統,并且由此可以修改caco3向ha的轉化過程(
最近,本發明人成功地制備了基于非晶形聚磷酸鹽(聚p)的微粒(müllerweg,etal.anewpolyphoshatecalciummaterialwithmorphogeneticactivity.materialsletters2015;148:163-166;müllerweg,etal.retinolencapsulatedintoamorphousca2+polyphoshatenanospheresactssynergisticallyinmc3t3-elcells.eurjpharmbiopharm2015;93:214-223),該微粒不僅在成骨細胞系統上以合成代謝方式起作用,而且還為它們的靶細胞(如成骨細胞)提供代謝燃料,并引起細胞內atp水平提高以及線粒體數量的增加(müllerweg,etal.amorphousca2+polyphoshatenanoparticlesregulatetheatplevelinbone-likesaos-2cells.jcellsci2015;128:2202-2207)。本發明人還在gb1420363.2中公開了這種生物相容性的、生物可降解的和具有生物活性的基于聚p的材料。在gb1420363.2中描述的微粒的大小,可以通過確定的pi:ca2+摩爾比來調節(müllerweg,etal.(2015)anewpolyphoshatecalciummaterialwithmorphogeneticactivity.materialslett148:163-166)。所形成的顆粒保持非晶形狀態,因此容易被堿性磷酸酶(alp)酶促水解。
聚p在血液中以相當大的量存在并且較大程度存在血小板中,并且已經被認為是用于形成骨磷酸鈣沉積物的磷酸鹽來源。通過alp從該聚合物酶促去除正磷酸鹽,其可以用作骨礦化的供體。此前,發明人描述了聚p調節細胞特異性分化過程,例如用模型細胞系saos-2細胞將礦物沉積物形成到骨形成成骨細胞上,使用模型細胞系raw264.7,誘導alp以及使opg(骨保護素):rankl(核因子κb配體的受體激活劑)的比例向合成代謝的成骨細胞途徑改變,并由此抑制破骨細胞的功能。此外,聚p誘導用于編碼骨形態發生蛋白-2(bmp-2)和支架結構纖維所成的系統膠原蛋白的基因。酶介導的骨形成中的現有技術和聚p的作用已被描述于,例如:
wangxh,
wangxh,etal.(2015)polyphoshateasametabolicfuelinmetazoa:afoundationalbreakthroughinventionforbiomedicalapplications.biotechnolj.doi:10.1002/biot.201500168;
müllerweg,etal.(2015)non-enzymatictransformationofamorphouscaco3intocalciumphosphatemineralafterexposuretosodiumphosphateinvitro:implicationsforinvivohydroxyapatiteboneformation.chembiochem16:1323-1332;和
müllerweg,etal.polyphoshate:amorphogeneticallyactiveimplantmaterialservingasmetabolicfuelforboneregeneration.macromolecbiosci2015;15:1182-1197。
因為現有材料的局限性(例如患者的長期固定期,植入物引起的感染等),迫切需要新的骨植入材料。理想地,這些植入物必須遵循骨形成的自然過程的原理,允許受損骨組織的快速再生。
在本發明的一個方面,發明人描述了聚p可以穩定acc相。在本發明的方法中,在5%[w/w]的水平下,聚p顯著抑制acc轉化為結晶caco3,在10%[w/w]的百分比下,該聚合物幾乎完全阻斷了該過程。這個發現是意想不到的。以前已經報道,可以將摻有規定ca2+摩爾比的可溶性na-聚p加工成固體納米尺度的納米顆粒/微粒,其保持非晶形(müllerweg,etal.(2015)anewpolyphosphatecalciummaterialwithmorphogeneticactivity.materialsletters148:163-166;müllerweg,etal.(2015)retinolencapsulatedintoamorphousca2+nanospheresactssynergisticallyinmc3t3-e1cells.eurjpharmbiopharm93:214-223)。不能期望,ca-聚p可以作為亞穩態acc的穩定劑;另參見:gb1420363.2和gb1502116.5。
聚p作為骨細胞上的具有形態發生活性的無機分子,誘導其礦化(gb1420363.2、gb1502116.5、gb1403899.6)。在本申請中,發明人還顯示含有5%[w/w]或10%[w/w]聚p的caco3,在saos-2細胞以及人間充質干細胞(msc)中通過誘導alp和骨形態發生蛋白2(bmp2)的基因表達而包含成骨潛能。更令人驚奇和意外的是,acc以“協同”方式增強了聚p對bmp2表達的刺激作用。此外,本發明人證明,本發明的acc/聚p混合材料是生物相容性的并且在體內支持再生,使其成為用于骨替換/植入物的有希望的支架材料。
在本發明的另一方面,本發明人使用用于從氯化鈣和磷酸氫二銨開始制備cap的技術。在新穎的方法中,除了用10:6的特征性ca/p摩爾比制備ha之外,他們還制備了混合有各種量聚p的cap。發明人意外地發現,含有>10重量%聚p的cap制品(相對于改性cap/ha沉積物)是非晶形的,而含有<10重量%聚p的cap/ha樣品由結晶相組成。發現所有樣品都支持骨細胞相關saos-2細胞的生長,但令人驚奇的是,只有含有10重量%(wt.%)的聚p的cap制品引起強的形態發生活性,如通過測量用于編碼alp和i型膠原蛋白的基因(骨和骨相關細胞分化的標記基因)表達所確定的。基于這些結果,本發明的基于聚p/cap的材料可能有益于作為骨替代植入物的應用。
此前,本發明人開發了在規定濃度cacl2存在下于環境條件下制備的基于聚p的材料。該材料由生物相容性的和生物可降解的球形非晶形顆粒組成。
現在發明人驚奇地發現,可將這種以微粒范圍尺寸制備的具有生物活性的材料用于通過形成到硅烷偶聯劑aptms的ca2+離子橋接的ti-6a1-4v支架的表面包被層,如電子顯微鏡和元素分析(edx)方法所證明。該發現和包被層的高耐久性和穩定性是出乎意料的,尤其是由于ca-聚p顆粒的微粒性質。
ca-聚p包被的鈦合金的另一令人驚奇的特性是,其作為骨樣saos-2細胞生長的合適基底的特性,盡管其明顯降低的表面粗糙度不可能支持細胞附著,甚至更多,與未處理的鈦支架相比,其誘導這些細胞表達關鍵酶(碳酸酐酶ix(caix)和alp)的能力,所述酶參與啟動酶誘導的骨礦物質沉積。
基于它們的特性,本發明的包被的鈦支架是用于制備具有新穎的再生誘發特征的高精度植入物的有希望的材料,其可以以個體化的尺寸和形狀生產。
眾所周知,na-聚p容易螯合ca2+并形成不溶性沉淀。因此,可以預期,向cacl2和(nh4)2hpo4中加入na-聚p將導致非晶形ca-聚p和結晶cap(ha)礦物沉積物的共沉淀。
在本發明的另一方面,本發明人在沉淀過程中,與cacl2和(nh4)2hpo4(用于在水溶液中形成ha的底物)一起加入了增加濃度的na-聚p。令人驚奇和意外的是,他們發現,在同時制備尺寸約為100nm的非晶形聚p/cap混合顆粒的情況下,10wt.%的聚p的含量防止了結晶ha的形成。以本發明這個方面為基礎的結果總結如圖1所示。
這一發現是重要的,因為由于不利的物理(低溶解度)和機械特性(脆性),即使是生物相容性的結晶ha,其應用目前也僅限于粉末、包被層和多孔體以及非承重植入物(wangmc,etal.crystallinesize,microstructureandbiocompatibilityofhydroxyapatitenanopowdersbyhydrolysisofcalciumhydrogenphosphatedehydrate(dcpd).ceramicsintern2015;41:2999-3008)。如果cap可以在非晶形相中合成,則可以避免這些缺點。
本發明人證明,非晶形聚p/cap顆粒(縮寫:acap-聚p)結合細胞生長促進活性顯示出不同的形態發生活性。他們發現acap-聚p引起兩個用于骨形成的標記基因(i型膠原蛋白和alp)的強烈上調。acap-聚p的效力與ca-聚p相當。
基于其引發形態發生活性的特性,本發明的acap-聚p顆粒提供了用作人造骨植入物的有希望的材料,其由生理代謝物/聚合物制備。
在本發明的另一方面,本發明人驚奇地發現,可通過聚p來穩定亞穩態acc相。在人骨中,acc形成為結晶碳酸化磷灰石/ha的前體。聚p用作非酶促碳酸鹽/磷酸鹽交換的磷酸鹽源。本發明人證明,關于caco3,在5%[w/w](稱為“ccp5”)的百分比時,聚p抑制acc向結晶caco3轉變,而在10%[w/w](稱為“ccp10”)時幾乎完全抑制。他們顯示兩種制品都是非晶形的,但也含有少量的球霰石,如通過xrd、ftir和sem分析所顯示的。
本發明人證明,與方解石相反,本發明的acc/聚p顆粒顯示成骨活性。它們誘導saos-2細胞以及人間充質干細胞(msc)中編碼alp的基因的表達,以及bmp2基因的表達。此外,在大鼠的體內研究中,使用含有本發明的acc/聚p材料并插入動物背部肌肉的plga微球體,本發明人證明包封的acc/聚p顆粒不僅是生物相容性的,而且還支持植入區域的再生。令人驚奇的是,與生物惰性方解石相比,含有少量球霰石的acc具有成骨潛力和優異的特性。基于這些特性,本發明的材料代表用于骨植入物的有希望的支架材料。
以下關于聚p的專利申請被認為是相關的:gb1406840.7、gb1403899.6、wo2012/010520、gb1420363.2、gb1502116.5和gb1510772.5。
在gb1420363.2中,本發明人公開了制備由鈣-聚p微粒組成的材料的方法,該材料顯示出以下特性:(i)它是非晶形的,和(ii)在能夠礦化的細胞系統中具有生物活性。
結果也報道于:müllerweg,etal.anewpolyphosphatecalciummaterialwithmorphogeneticactivity.materialsletters2015;148:163-166;müllerweg,etal.retinolencapsulatedintoamorphousca2+polyphosphatenanospheresactssynergisticallyinmc3t3-e1cells.eurjpharmbiopharm2015;93:214-223;和müllerweg,etal.polyphosphate:amorphogeneticallyactiveimplantmaterialservingasmetabolicfuelforboneregeneration.macromolecbiosci2015;15:1182-1197。
在gb1420363.2中描述的材料的特性,優于ha(也參見實施例)以及用于骨再生和修復的常規聚p制品(例如gb1406840.7和gb1403899.6)。
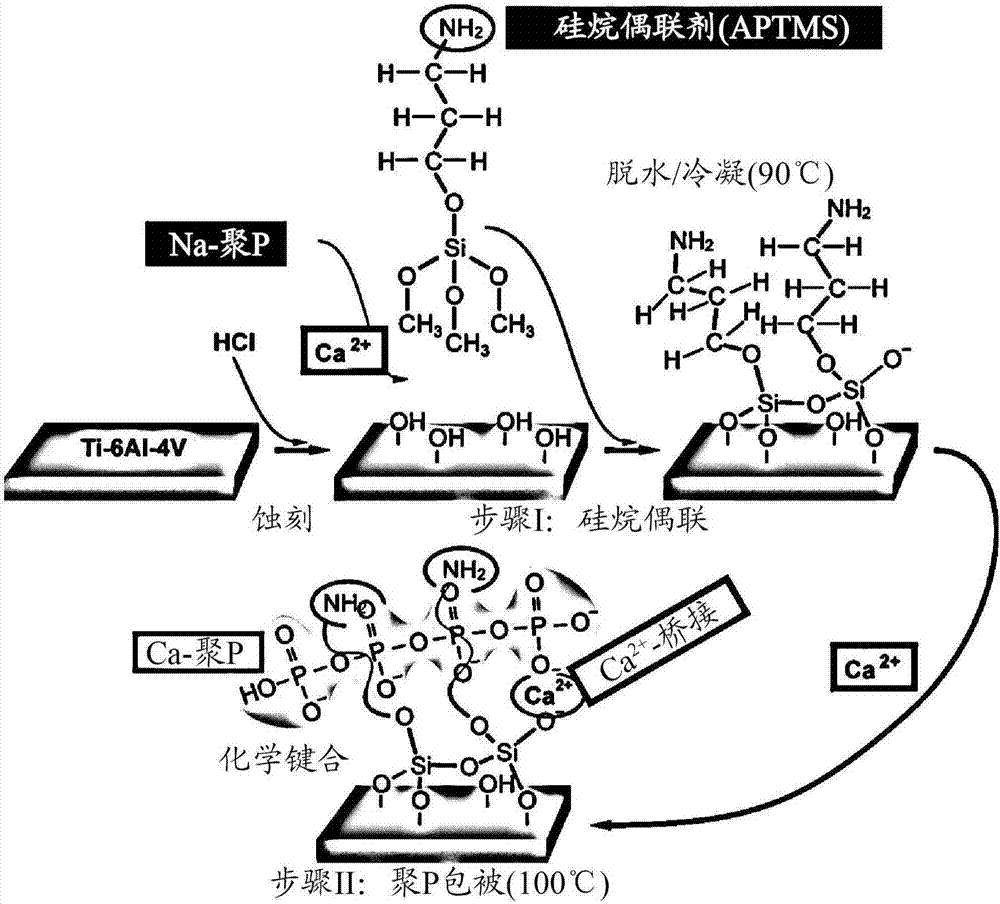
現在,本發明人成功地開發出通過其可以用聚p緊密地覆蓋鈦/鈦合金的步驟。在用hcl蝕刻后,金屬表面與aptms共價連接,之后ca-聚p顆粒可以通過ca2+離子鍵連接到所述表面(圖2)。
在金屬表面的本發明的聚p包被,證明是耐用的并且令人驚奇地穩定。
aptms可以被其它硅烷偶聯劑代替,例如3-(三甲氧基硅基)丙基甲基丙烯酸酯。除了聚p之外,aptms另外的官能團允許肽與硅烷包被的鈦表面結合。
與未處理的鈦圓片相當高的表面粗糙度(最大約為7μm)相比,ti-ca-聚p圓片是平滑的,最大粗糙度為0.8μm。通常,與中等粗糙表面相比,細胞附著到非常光滑表面的程度較低(例如,huanghh,etal.effectofsurfaceroughnessofgroundtitaniumoninitialcelladhesion.biomoleng2004;21:93-97)。因此,出乎意料的是,在沒有任何圓片的對照試驗中看到,聚p包被的圓片允許saos-2細胞以一定速率生長。
發明人顯示,在2天的孵育期后,在含有未處理的鈦圓片試驗中的細胞死亡。這與在這段時間內saos-2細胞密集地附著到ti-ca-聚p圓片并形成幾乎均勻的單細胞層的觀察結果形成鮮明對比。令人驚訝的發現是,在ti-ca-聚p圓片上生長的細胞顯示細胞擴散的表型形態,這是活性細胞代謝和細胞遷移的明顯標志。
“關于”表示所示值的+/-10%。
本發明的另一方面涉及以下發現:本發明的ca-聚p包被層能夠刺激骨形成細胞的功能活性,如通過在包被的金屬表面(與未處理的鈦表面相比)上生長的細胞中編碼碳酸酐酶ix(caix)和alp的轉錄物的增加的穩態水平所證明(如用qrt-pcr定量的)。
酶ca參與骨形成的引發(形成caco3沉積物;müllerweg,etal.inductionofcarbonicanhydraseinsaos-2cells,exposedtobicarbonateandconsequencesforcalciumphosphatecrystalformation.biomaterials2013;34:8671-8680;wangxh,etal.enzyme-basedbiosilicaandbiocalcite:biomaterialsforthefutureinregenerativemedicine.trendsbiotechnol2014;32:441-447)。
alp是具有功能活性的礦物沉積而形成成骨細胞的確定標志物(參見:wangxh,etal.bio-silicaandbio-polyphosphate:applicationsinbiomedicine(boneformation).curropinbiotechnol2012;23:570-578)。
本發明的數據顯示鈦氧化的ti-6a1-4v支架是用于體外骨樣saos-2細胞的惰性基底。如果包被具有形態發生活性的ca-聚p聚合物,則該金屬獲得生物功能特性。具有聚p的這種植入材料的生物功能化的進展,不僅提供個體化植入物的制備,而且還提供了將硬而脆的金屬植入物的機械特性與較軟的骨骼及其周圍組織的機械特性相匹配的有利特性。
聚p的鏈長可以在約3個至約1000個磷酸鹽單元的范圍,優選在約20個至約200個磷酸鹽單元的范圍,以及最優選約40個磷酸鹽單元。
本發明方法中使用的ca-聚p微粒的優選組成是化學計量比(磷酸鹽與鈣)為:0.1比1至50比1,優選1比1至10比1,以及最優選7比1。
盡管它們的直徑(0.2μm和3μm)在允許受體介導的胞吞作用(約50nm)的范圍之外,出人意料的是,ca-聚p微粒是具有生物活性的。
與通過常規技術制備的ca-聚p鹽相比,該聚p材料是生物可降解的,并且顯示出優異的形態發生活性。
本發明方法的另一方面是,應用/使用該方法來制備具有生物活性的鈦合金植入物。本發明方法的另一方面是,應用/使用該方法來制備刺激成骨細胞活性的植入物。本發明方法的另一方面是,將ca-聚p包被的鈦合金表面和植入物與鎵鹽組合應用/使用,以便利用它們在通過應用本發明方法所制備的包被層上的增強的協同效應。發明人意外地發現,鎵鹽(如硝酸鎵)增強了具有生物活性的ca-聚pti合金包被層對具有功能活性特征的成骨細胞的轉錄物的表達、穩態水平的刺激作用。這一發現令人驚奇,因為已經報道鎵鹽只能通過破骨細胞調節骨吸收,但不影響或僅最低限度影響成骨細胞的基因表達和alp活性(verrone,etal.galliummodulatesosteoclasticboneresorptioninvitrowithoutaffectingosteoblasts.brjpharmacol2010;159:1681-1692)。
本發明的另一方面涉及令人驚奇的發現,即如果在通常導致結晶ha合成的步驟中,聚p以一定濃度存在,也可以制備與結晶ha相比具有優異特性,并且實現與gb1420363.2中公開的聚p微粒幾乎相同的生物活性(形態發生活性、刺激骨相關基因表達)的含有非晶形聚p的材料。
由本發明人開發的制備具有生物活性的非晶形聚p取代的cap顆粒(acap-聚p)的本發明方法,包括以下步驟:
a)將聚p鹽的水溶液加入到磷酸鹽源的水溶液中;
b)將所得溶液加入到溶解的鈣鹽中;
c)將ph調節至堿性值,優選約10;和
d)收集、洗滌和干燥數小時后,優選在室溫下24小時后形成的所得沉淀。
聚p鹽優選為聚磷酸鈉(na-聚p)。如果聚p鹽的量高于5wt.%的聚p鹽,則形成本發明的聚p-取代的cap顆粒(acap-聚p),稱為cap制品。作為實例,用10wt.%的聚p鹽的聚p取代的cap顆粒(acap-聚p)已經實現了最佳結果。
根據本發明方法制備的形成本發明聚p取代的cap顆粒(acap-聚p)的cap成分的鈣鹽和磷酸鹽源,可以分別是氯化鈣(cacl2)和磷酸氫二銨[(nh4)2hpo4]]。
通過本發明方法制備的聚p取代的cap顆粒(acap-聚p)獲得最佳結果,其中計算鈣鹽的量和用作磷酸鹽源的試劑的量以獲得ca/p摩爾比為10:6的cap。
聚p取代的cap顆粒(acap-聚p)的平均尺寸可以在約20nm至約300nm的范圍,優選在約70nm至約120nm的尺寸范圍。
本發明的另一方面涉及以下發現:本發明的聚p-取代的cap顆粒(acap-聚p)能夠刺激骨形成細胞的功能活性,如通過在骨形成saos-2細胞中編碼i型膠原蛋白(col-i)和堿性磷酸酶(alp)的轉錄物的增加的穩態水平所證明(如用qrt-pcr定量的)。
意外地是,這些聚p取代的cap顆粒(acap-聚p)具有生物活性,盡管它們的直徑(70-120nm)大于可被受體介導的內吞作用(約50nm)攝取的顆粒的直徑。
與通過常規技術制備的ha晶體相比,聚p取代的cap顆粒(acap-聚p)是生物可降解的,并且顯示出優異的形態發生活性。
本發明方法的另一方面是,應用該方法制備生物活性植入材料。本發明方法的另一方面是,應用該方法來制備刺激成骨細胞活性的人造骨植入物。
本發明的另一方面涉及制備含有少量球霰石的acc多晶型物。發明人在合成acc期間,將陰離子聚合物聚p的na+鹽加入到caco3(cacl2和na2co3)的前體中(圖3)。令人驚奇的是,本發明人發現,在最終濃度為10%時聚p幾乎完全防止acc轉變為其結晶多晶型物球霰石、霰石和方解石。
分別測試的caco3固體和聚p生理代謝物都具有成骨潛力,并且可以作為具有生物活性的骨移植物的成分。反過來,所開發的支架不僅利用聚p的形態發生潛力,而且還利用該聚合物的特性在acc階段冷凍caco3固體。該材料的成骨活性優于方解石;它通過刺激成骨細胞來強烈誘導編碼alp(骨形成標志物)的基因的表達。該結果已經從骨樣saos-2細胞和msc的研究中獲得。
此外,發明人證明,acc/聚p通過成骨細胞強烈上調bmp2(骨形成誘導物)的表達。更重要的是:他們驚奇地發現acc以“協同”的方式增加由聚p誘導的bmp2表達,導致bmp2轉錄水平的更快上升。可以預期本發明的acc/聚p微粒的這種效應將導致骨缺損的更快愈合。
acc/聚p材料不僅具有生物相容性,而且還支持受損植入區域的細胞再生。為了評估acc/聚p材料在體內的生物相容性,本發明人將本發明的材料包封在plga微球體中。并行地,對照球體保持沒有acc/聚p。將珍珠/微球嵌入大鼠背部的肌肉。觀察期2周、4周和8周后,從大鼠中取出組織樣本,并在切片和用mayer蘇木精染色后進行顯微鏡檢查。檢查顯示,在具有含有acc/聚p材料的微球的動物中,分別在4周和8周后,具有細胞的植入物區域的晚期增殖(repopulation)變得明顯。相反,缺少acc/聚p的微球沒有任何細胞。通過測量植入區域組織樣本的硬度(中值redym剛度)來支持這些結果,其顯示了在8周時間段后與對照相比顯著增加1.8倍(植入前在肌肉樣品中測量值的81%)。
本發明人開發的用于制備本發明的acc/聚p材料的優選方法,包括以下步驟:
a)在例如1升0.1mnaoh中制備含有聚p鹽的溶液;
b)向該溶液中加入0.5mol/l的na2co3;
c)用1.5倍體積的去離子水稀釋所得溶液;
d)將該溶液與相同體積的含有0.5mol/lcacl2·2h2o的水溶液快速混合(導致鈣離子和碳酸根離子之間的等摩爾濃度比);和
e)在室溫下用丙酮洗滌后,過濾并干燥沉淀物。
用于制備本發明的acc/聚p微粒的0.1mnaoh溶液中聚p鹽的優選濃度,在0.001mol/l至1.0mol/l的范圍,優選在0.01mol/l至0.1mol/l的范圍(基于磷酸鹽單位)。
如果用于制備本發明的acc/聚p微粒的0.1mnaoh溶液中的聚p鹽的濃度為0.025mol/l,或者,甚至更優選為0.05mol/l(基于磷酸鹽),則獲得最佳結果。所得的制品分別稱為“ccp5”和“ccp10”。聚p鹽優選為na-聚p。
聚p的鏈長可以在3個至約1000個磷酸鹽單元的范圍。具有平均鏈長為約10個至約100個磷酸鹽單元的聚p分子獲得最佳結果,并且在該范圍內最佳約40個磷酸鹽單元。
本發明的另一方面涉及以下發現:本發明的acc/聚p顆粒通過在骨形成saos-2細胞中(如用qrt-pcr定量的)誘導編碼alp和bmp2的基因表達而顯示出成骨活性。
與不存在聚p下由acc快速形成的方解石相比,acc/聚p顆粒是生物可降解的,并且顯示出優異的形態發生活性。
發明人證明,使用具有10%[w/w]聚p(“ccp10”)的acc制品,ca2+和co32-的同時釋放在孵育的最初48小時期間是快速的,從而允許從支架釋放生物學活性陰離子co32-和po43-。如本發明人之前所證明的那樣,正磷酸鹽將從聚p中酶促地釋放出來(müllerweg,wangxh,diehl-seifertb,kropfk,schloβmacheru,lieberwirthi,glasserg,wiensm,
本發明方法的另一方面是,應用該方法制備具有生物活性的植入材料。本發明方法的另一方面是,應用該方法來制備刺激成骨細胞活性的人造骨植入物。此外,本文描述的本發明的另一方面是,通過應用本發明方法來制備的植入物。
用聚p穩定亞穩態acc的本發明方法,還允許將acc/聚p顆粒應用于膳食補充劑。如發明人所證明的,例如,參見圖18,這些顆粒,例如,與結晶方解石多晶型物相比,“cc10”在長時間的孵育期內釋放鈣。
因此,本發明的acc/聚p顆粒也可以用作治療鈣缺乏癥的膳食補充劑。
因此,本發明的另一方面是,穩定的acc(acc/聚p)作為膳食補充劑用于預防/治療骨質疏松癥的用途。
鈣在許多生物過程(例如細胞內信號轉導、肌肉收縮、神經元傳遞和血管收縮/血管擴張)中起重要作用。通過聚p穩定的acc可以作為鈣的易于獲得的食物補充劑,用于預防/治療與鈣代謝紊亂相關的許多病理學病癥。
根據最近關于生物種子caco3性質的發現,可以在骨形成過程中描述通過po43-進行co32-的陰離子交換和從聚p供給正磷酸鹽,接下來一系列的機制不同的過程(圖4)。在骨礦物質沉積的第一階段,像在軟骨內骨化中,使包含長骨的骨骺和骨干之間的生長中心的干骺端中的軟骨鈣化。很可能是由ca-ii和/或ca-ix來酶促驅動這種鈣化過程。第二,除了在骨形成區域和骨折部位的成骨細胞之外,積聚的血小板將聚p釋放到細胞外空間,其中聚合物在正磷酸鹽釋放下經歷alp介導的胞外水解。第三,在生物種子合成的空間附近形成的可用的磷酸鹽單元,作為形成acp的來源。如本方案中所描繪的,本發明的材料是有希望的生物相容性成骨支架,其提供用于生物種子開發(caco3[co32-])和隨后轉化為磷酸鈣(聚p[po43-])的底物。
因此,總之,本發明涉及制備具有生物活性的鈦合金包被層的方法,其包括以下步驟:a)通過將聚磷酸鈉(na-聚p)的水溶液與二水氯化鈣(cacl2·2h2o)水溶液在形成膠體懸浮液的條件下,在升高的溫度,優選在90℃下混合幾小時,優選3小時來制備聚磷酸鈣(ca-聚p)微粒;b)使用硅烷偶聯劑將所述ca-聚p微粒膠體懸浮液偶聯到合適的鈦合金支架上;和c)將b)的懸浮液的ph值調節至微堿性值,優選為8.0,以允許通過ca2+離子鍵形成將聚磷酸鹽(聚p)結合到硅烷官能化的金屬支架。鈦合金可以是ti-6a1-4v。硅烷偶聯劑可以是(3-氨丙基)三甲氧基硅烷[aptms]。
本發明還涉及用于制備具有生物活性的非晶形聚磷酸鹽取代的磷酸鈣顆粒(“acap-聚p”)的方法,其包括以下步驟:a)將聚磷酸鹽的水溶液加入到磷酸鹽源的水溶液中;b)將所得溶液加入到溶解的鈣鹽中;c)將ph調節至堿性值,優選10;和d)收集、洗滌和干燥數小時后,優選在室溫下24小時后形成的所得沉淀。聚磷酸鹽可以是聚磷酸鈉。
本發明還涉及用于制備具有生物活性的非晶形碳酸鈣(acc)-聚磷酸鹽微粒的方法,其包括以下步驟:a)制備在約0.1m氫氧化鈉中的聚磷酸鹽水溶液;b)向所述溶液中加入約0.5mol/l的碳酸鈉;c)用約1.5倍體積的去離子水稀釋所得溶液;d)將所述溶液與相同體積的含有氯化鈣的水溶液混合,從而產生鈣離子和碳酸根離子之間的約等摩爾濃度比;e)在約室溫下用諸如丙酮的低級烷基酮洗滌;和f)過濾和干燥形成的沉淀物,基于磷酸鹽,步驟a)中聚磷酸鹽的濃度在約0.001mol/l至約1.0mol/l的范圍,優選在約0.01mol/l至約0.1mol/l的范圍。優選地,基于磷酸鹽,步驟a)中聚磷酸鹽的濃度為約0.025mol/l或約0.05mol/l。
優選的方法是,其中聚磷酸鹽的鏈長在約3個至約1000個磷酸鹽單元的范圍,優選在約10個至約100個磷酸鹽單元的范圍,以及最優選約40個磷酸鹽單元。優選的方法是,其中聚磷酸鹽的量高于磷酸鈣制品的5wt.%,優選10wt.%。
優選的方法是,其中鈣鹽是氯化鈣(cacl2),磷酸鹽源是磷酸氫二銨[(nh4)2hpo4]]。
在本發明中,聚磷酸鈣微粒的特征在于:磷酸鹽比鈣的化學計量比為0.1比1至50比1,優選1比1至10比1,或化學計量比為7比1。
優選的方法是,其中計算所述鈣鹽的量和用作磷酸鹽源的試劑的量,以獲得ca/p摩爾比為10:6的磷酸鈣。優選的方法是,其中聚磷酸鈣微粒的平均尺寸在約0.1μm至約30μm,優選0.8μm至3μm的范圍。優選的方法是,其中聚磷酸鹽取代的磷酸鈣顆粒(“acap-聚p”)的平均尺寸為約20nm至約300nm,優選約70nm至約120nm的范圍。
優選的方法是,其還包括制備具有生物活性的鈦合金植入物的步驟。還優選的方法是,其還包括制備具有生物活性的植入材料的步驟。還優選的方法是,其還包括將至少一種鎵鹽包含在所述植入物中的步驟。優選地,所述具有生物活性的植入材料是人造骨植入物。優選地,所述具有生物活性的植入材料是人造骨植入物。
本發明還涉及通過本發明的方法制備的植入物,或通過本發明的方法制備的穩定的acc組合物。
優選地,根據本發明制備的包被層可以用作植入物(任選地與至少一種鎵鹽組合使用),或作為食物或膳食補充劑(例如acc組合物)。穩定的acc組合物用于治療鈣缺乏癥,或用于預防和/或治療骨質疏松癥。
現在將在以下優選實施例中進一步描述本發明,但并不受限于此。為了本發明的目的,本文引用的所有參考文獻通過引用整體并入。
附圖說明
圖1顯示從前體ca2+、po43-和oh-形成非晶形cap(acap)的示意圖。acap經歷了成熟為結晶ha,或者在<10wt.%的聚p存在下同樣成熟為結晶cap(參見底部的插圖,顯示cap晶體;sem圖像)。如果cap沉淀物中聚p的含量增加至>10wt.%,則形成球狀非晶形acap-聚p(參見頂部的插圖;sem圖像)。
圖2顯示使用硅烷偶聯劑aptms將聚p結合到鈦圓片的示意圖。用hc1蝕刻鈦合金ti-6a1-4v,暴露于鈦圓片上的羥基與硅烷偶聯劑aptms交聯,所述偶聯劑形成至聚p的ca2+橋。脫水/縮聚后的偶聯劑仍然含有游離的反應性胺基,其可用于進一步偶聯至活性組分,例如,通過1-乙基-3-(3-二甲基氨丙基)碳二亞胺。在此過程中,金屬表面與硅烷共價連接,而硅烷又可以通過ca2+離子橋結合聚p。
圖3顯示制備方解石和補充有聚p的caco3的示意圖。插圖顯示各自產物的sem圖像。
圖4顯示軟骨內骨化過程和推薦的骨礦物質沉積階段的示意圖。穿透血管后,在骨干中的初級骨化中心的透明軟骨開始鈣化。在骨骺的次級骨化中心,松質骨的形成稍后開始。在骨骺和骨干之間的骨骺和骺板的表面(生長區),透明軟骨的兩個區域保留關節軟骨。生長區的礦物沉積被細分為i期:由膜相關ca-ix介導形成的非晶形碳酸鈣(acc)生物種子;ii期:在形成用于碳酸鹽-磷酸鹽轉移反應的正磷酸鹽情況下,從血小板釋放的聚p經歷alp介導的水解作用;和iii期:磷酸鹽單元用于(碳酸化)磷酸鈣形成。
圖5顯示與ti-ca-聚p圓片(b、d、f)相比,鈦合金圓片(a、c、e)的表面粗糙度。通過光學顯微鏡可視化圓片的表面,并使用vk分析儀軟件分析粗糙度。顯示了線掃描的軌跡(c、d)。在e、f中顯示代表性區域的高度剖面圖;數字表示法向量直線內的偏差的最大尺寸。
圖6顯示通過edx光譜(a、c、e)和sem(b、d、f),對鈦和ti-ca-聚p圓片進行的元素組成分析。(a、b)未處理的圓片(ti6a14v);(c、d)在聚p和cacl2反應試驗中,用較低濃度的aptms(1mg/試驗;聚p@ti6a14v-1)制備的ti-ca-聚p圓片;和(e,f)ti-ca-聚p圓片,其在較高aptms濃度(2mg/試驗;聚p@ti6a14v-h)存在下被包被。
圖7顯示鈦圓片對saos-2細胞生長的影響。將細胞在其他相同條件下接種到不含有鈦圓片(空白柱)、鈦合金圓片(交叉條紋柱)或ti-ca-聚p圓片(填充柱))的24孔板中。在1d、2d和3d的孵育期后收獲細胞,并通過xtt試驗確定細胞的存活力。數據表示10次獨立實驗的平均值±sd(*p<0.01)。
圖8顯示編碼(a)caix和(b)alp的基因的表達。將表達值標準化為gapdh的表達。在沒有任何鈦圓片(空白柱),或者在鈦合金圓片上(交叉條紋柱)或在ti-ca-聚p圓片上(填充柱)培養細胞。首先在mac不存在下孵育培養物3d;然后將它們轉移到補充有mac的培養基中,并且如所述,繼續孵育另外3d或5d。然后收集細胞,提取rna并進行qrt-pcr,以測定caix和alp轉錄物;將gapdh的表達作為參考。數據表示為五次獨立實驗的平均值±sd;每項實驗一式兩份進行(*p<0.01)。nd表示未檢出。
圖9顯示具有形態發生活性的ca-聚p的鈦圓片的包被層。如果包被了具有形態發生活性的ca-聚p聚合物,則金屬材料(ti-6a1-4v)獲得生物功能特性。在該過程中,鈦表面被蝕刻,導致羥基的暴露。在ph8時,它們與硅烷偶聯劑(例如aptms)形成共價連接。在這種環境下,在硅烷和聚p之間形成了ca2+離子橋。那些包被的鈦圓片允許骨樣saos-2細胞沉降并誘導它們進行基因表達(caix和alp);這些酶至關重要地參與骨礦物質/羥基磷灰石(ha)沉積。
圖10顯示從純na-聚p“聚p”和純“ha”以及在不同量的na-聚p存在下制備的ha(即“ha(2.5)聚p”中為2.5wt.%,“ha(5)聚p”中為5wt.%,和“ha(10)聚p”為10wt.%)獲取的衍射圖案。從底部到頂部給出各自的圖案。“聚p”和“acap(10)聚p”沒有看到衍射信號。突出顯示ha或結晶cap特征的衍射峰(■)。
圖11顯示“聚p”樣品和“ha”樣品以及cap樣品(其中正磷酸鹽已被聚p部分取代,“ha(2.5)聚p”樣品、“ha(5)聚p”和“acap(10)聚p”樣品)的ftir光譜。標記了“co32-和po43-”的一些振動帶;另外指出了h2o和co2帶的區域。(a)在4000至500的波數范圍(cm-1);(b)2000cm-1至500cm-1部分的放大。
圖12顯示聚p和cap顆粒的tem顯微照片。(a)“ha”晶體;(b和c)“ha(2.5)聚p”晶體和“ha(5)聚p”晶體;和(d)“acap(10)聚p”非晶形球狀顆粒。
圖13顯示在saos-2細胞中編碼(a)ⅰ型膠原蛋白(col-i)或(b)堿性磷酸酶(alp)的基因的穩態表達水平。將細胞暴露于10μg/lml的聚p納米顆粒“aca-聚p-np”(填充柱),或暴露于100mg/ml的“ha”(空白柱),“ha(2.5)聚p”(右陰影柱),“ha(5)聚p”(左陰影線柱)或“acap(10)聚p”(交叉陰影線柱)。在標準培養基/血清中初始孵育3d后,將細胞轉移到缺乏(減去mac)或含mac(加上mac)的培養基/血清中。在7d的孵育期后,收獲細胞,提取rna,隨后用于qrt-pcr分析。表達值以與參考基因gapdh的比值給出。結果是5次平行實驗的平均值;*p<0.01。
圖14顯示方解石以及“ccp5”(0.05gna-聚p/試驗)和“ccp10”(0.1gna-聚p))的ftir光譜。將方解石與acc的主要區別振動區域/信號,o-h(約3250cm-1)的振動范圍和在725cm-1處的co3的不對稱v2線加畫圓圈。
圖15顯示從(a)方解石和(b)含有兩種不同濃度的聚p的兩種caco3制品(“ccp5”或“ccp10”)獲得的xrd圖案。特征信號被突出顯示并用各自的米勒指數標記,在括號中給出。請注意在(a)和(b)之間的縱坐標標注的不同比例。
圖16顯示由cacl2·2h2o和na2co3形成的固體的形態;sem分析。(a和b)在不存在聚p的情況下,形成方解石晶體。在沉淀過程中加入聚p后,形態發生變化。(c和d)在存在5%聚p的情況下,“ccp5”顆粒顯示出球形外觀。(e和f)在10%聚p的情況下,“ccp10”,固體顯示出對應于球霰石晶體(vat)的薄片狀形狀。
圖17顯示在3d孵育期后,在50μg/ml的“ccp10”(a和b)或方解石(c和d)存在下,saos-2細胞的生長方式。通過相位差/nomarski光學鑒定細胞。試驗中的caco3顆粒在相差圖像中變得可見,并被標記(><)。
圖18顯示從caco3顆粒釋放ca2+。將“ccp10”或方解石在tris-hcl緩沖液(ph7.4)中孵育不同時間,分析上清液中ca2+濃度。結果是6次平行實驗的平均值;*p<0.01。
圖19顯示在(a)saos-2細胞和(b)msc中alp基因的穩態表達水平。細胞保持在沒有任何caco3固體的情況下(對照),或暴露于50μg/ml的“ccp5”(左側陰影線柱)、“ccp10”(右側陰影線柱)或方解石(填充柱)。在mac不存在下的3d預孵育期之后,在mac不存在(減去mac)或暴露于mac(加上mac)的情況下,繼續孵育細胞。在7d孵育后收獲細胞,提取其rna并進行qrt-pcr分析。表達值以與參考基因gapdh的比值給出。結果是5次平行實驗的平均值;*p<0.01;根據加晶種期間在細胞中測量的表達來計算所述值。
圖20顯示在“ccp10”和聚p(ca2+復合物)存在下,在saos-2細胞中bmp2基因的穩態表達水平。細胞保持在沒有任何添加劑的情況下(對照),或暴露于50μg/ml的“ccp10”(右陰影線柱)、5μg/ml的聚p(ca2+絡合物;50μm磷酸鹽單元;交叉陰影線柱)或50μg/ml方解石(填充柱)。在mac不存在下的3d預孵育期之后,在mac存在下將細胞繼續孵育長達7天,并通過qrt-pcr分析bmp2表達。表達值以與參考基因gapdh的比值給出。結果是5次平行實驗的平均值;*p<0.01;根據加晶種期間細胞中測定的表達來計算所述值(第0天);#p<0.01(僅適用于“ccp10”);在相應的孵育期間,根據在具有聚p(ca2+絡合物)的細胞中測量的表達來計算所述值。
圖21顯示微球的形態;(a)對照球“cont-mic”,和(b)加載聚p的球體“聚p-mic”。
實施例
在以下實施例中,僅針對鏈長為40個磷酸鹽單元的聚p分子描述本發明的方法。通過使用具有較低和較高鏈長(如約20個至100個單元)的聚p分子可獲得類似的結果。
鈦-ca-聚p(ti-ca-聚p)圓片
鈦合金(ti-6a1-4v)圓片被蝕刻,以允許與硅烷偶聯劑aptms交聯(圖2)。在第二步,通過ca2+離子橋用聚p覆蓋圓片。最后將樣本ti-ca-聚p圓片在100℃下干燥。我們目的是使用硅烷偶聯劑aptms來提供另外的官能團(胺基團),以將生物活性肽偶聯至聚p包被的金屬表面。鈦圓片的功能化也用3-(三甲氧基硅基)丙基甲基丙烯酸酯成功地進行,僅允許聚p-鈦包被層。
鈦合金圓片與ti-ca-聚p圓片之間的比較(光學顯微鏡圖像)顯示,與鈦合金圓片的深灰色表面顏色相反,ti-ca-聚p圓片具有閃亮的銀白色外觀。在用聚p包被圓片的表面之后,它們失去其高的粗糙度。雖然未處理的圓片具有≈5.5μm的平均粗糙度,最大值為7.02μm(圖5a、圖5c、圖5e),但是聚p包被的圓片最大程度地暴露了0.78μm的表面粗糙度(圖5b、圖5d、圖5f)。
通過edx光譜進行鈦圓片表面的元素特異性分析(圖6)。未經處理的圓片的表面,在4.5kev顯示出鈦的優勢kα峰,并在4.9kev顯示出較低kβ峰(圖6a)。表面的形態被標記為粗糙(圖6b)。如果在有聚p和cacl2的包被溶液中孵育后,蝕刻并與較低濃度的aptms(1mg/試驗)反應后檢查鈦圓片,通過sem可解析ca-聚p微粒(müllerweg,tolbae,
ca-聚p包被層的耐久性
如下文“方法”部分所述,通過測定來自sbf(缺少ca2+作為組分)中包被圓片的ca2+釋放,來測量聚p的表面包被層。在平行試驗中,測量了來自ti-ca-聚p圓片以及未處理的鈦圓片(對照)的ca2+釋放。作為另外的對照,將一ti-ca-聚p圓片插入補充有1μg/ml的alp的sbf培養基中;將所有樣品于37℃孵育。在所有三種試驗中的時間為零時,ca2+濃度為<3μg/ml。在孵育培養基中1d后,測定的ca2+量如下:ti-ca-聚p圓片:<3μg/ml(<3μg/ml[對照];12±3μg/ml[ti-ca-聚p圓片+alp]);進行5次平行試驗。與和alp一起的ti-ca-聚p圓片的試驗相反,在3d的孵育期后,在含有ti-ca-聚p圓片的試驗中ca2+釋放略微增加。測量以下值:5.2±0.8μg/ml[ti-ca-聚p圓片](<3μg/ml[對照];86.9±3.2μg/ml[ti-ca-聚p圓片+alp])。在孵育試驗12d后,其值如下:9.7±1.2μg/ml[ti-ca-聚p圓片];<3μg/ml[對照];153.1±17.1μg/ml[ti-ca-聚p圓片+alp]。
saos-2細胞在鈦圓片上的生長
通過如下文“方法”部分中所述的xtt試驗,來測定骨樣saos-2細胞的總體生長速率。對于所有三個平行的實驗系列,將細胞以3×104個細胞/孔(2ml試驗)的密度接種;不含鈦圓片的試驗、鈦合金圓片試驗、ti-ca-聚p圓片試驗(圖7)。在1-d孵育期后,細胞密度從0.3吸光度單位增加到0.49±0.6單位(無圓片試驗)和0.47±0.05單位(含ti-ca-聚p圓片),而在有鈦合金圓片的試驗中密度下降至0.26±0.03單位。該趨勢在隨后的孵育期內增加,并且在3-d孵育期后所述值達到:0.09±0.02(鈦合金圓片;顯著降低)、0.72±0.08(無圓片)和0.68±0.07(ti-ca-聚p圓片)。這些數據意味著鈦表面不支持saos-2細胞的生長,而在ti-ca-聚p圓片上生長的細胞顯示與沒有任何圓片的培養物相同的生長動力學。
通過染色圓片表面,還強調了包被有聚p的圓片的特性,其是用于saos-2細胞生長的非常合適的基底。未包被聚p的鈦合金圓片與saos-2細胞孵育3d;在此之后,沒有在圓片上看到細胞。相比之下,如果將聚p包被的ti-ca-聚p圓片在saos-2細胞存在下孵育相同的時間段,則觀察到幾乎均勻的單細胞層。在較高放大率下仔細檢查顯示,細胞顯示出細胞擴散的特性,這是細胞至關重要的生存和生長的特征信號。
作為對saos-2細胞容易生長到ti-ca-聚p圓片上的結論的進一步支持,分析了覆蓋有細胞區域的元素碳(c)、鈦(ti)和磷(p)的分布。通過基于sem的edx映射,進行元素的半定量測定。通過記錄背散射電子獲得細胞的定位。在細胞生長區域內,測量元素c的高積聚,在細胞生長的圓片的周圍表面上,細胞區域外部的鈦和聚p顯著。
碳酸酐酶ix和堿性磷酸酶的表達
作為生長在鈦圓片上的saos-2細胞功能活性的標記物,通過定量qrt-pcr測定了編碼酶碳酸酐酶ix(caix)和堿性磷酸酶(alp)的兩個基因的表達。在mac不存在下生長3d的saos-2細胞中caix轉錄物的穩態水平的研究,對于含有鈦合金圓片的培養物,表達水平從0.31±0.03(在接種時間)到0.12±0.01顯著降低,而不存在圓片或存在ti-ca-聚p圓片的培養細胞中的水平分別從0.27±0.02和0.25±0.03增加到0.38±0.04和0.40±0.05(圖8a)。隨后在mac存在下孵育培養物,導致在不含圓片或ti-ca-聚p圓片浸沒的試驗中caix表達水平的增加。在mac存在下5d后,檢測到在不存在圓片的細胞中,caix轉錄物水平從0.38±0.04至0.81±0.08顯著增加。在ti-ca-聚p圓片上培養的細胞中也發現該基因表達從0.41±0.05至0.59±0.06顯著增加。這些結果強調,用聚p包被到鈦植入材料上提供了具有生物活性表面的材料(圖9)。
同時,同樣通過qrt-pcr來測定酶alp基因的表達。再次,數據(圖8b)顯示,在3d孵育期后,相對于不存在任何圓片,在含有鈦合金圓片的培養物中alp的表達水平顯著降低。稍后在孵育過程中,水平低至表達無法可靠地記錄。相比之下,在mac存在下alp的穩態表達,在無圓片的試驗中和在有ti-ca-聚p圓片的試驗中都顯著增加,分別為從0.038±0.005到0.097±0.007(在沒有圓片的第5天),從0.034±0.004到0.074±0.007。
在另一組實驗中,在100μm硝酸鎵存在下進行測定(參見表1)。如上所述,在沒有任何鈦圓片、或在鈦合金圓片上、或在ti-ca-聚p圓片上培養細胞,首先在mac不存在下培養3d,然后在補充有mac的培養基中再培養5d。結果表明,與硝酸鎵不存在下為0.27±0.02至0.81±0.08(另見圖8)相比,在圓片不存在下,在mac不存在下生長3d,以及隨后在mac存在下生長5d的saos-2細胞中,caix轉錄物的穩態水平從0.24±0.05增加至0.89±0.11(表1)。如果將細胞在ti-ca-聚p圓片上培養,發現與不存在硝酸鎵的試驗相比,在硝酸鎵存在下caix轉錄物的水平更強的增加;相對于從0.25±0.04至0.59±0.06(不存在硝酸鎵;還參見圖8),caix轉錄物的穩態水平從0.27±0.04增加到0.96±0.15(存在100μm硝酸鎵),表明ca-聚p包被層和鎵鹽有很強的協同效應(與沒有圓片和有ti-ca-聚p圓片的試驗比較)。
如果在不存在圓片和存在鈦合金圓片或ti-ca-聚p圓片的情況下,測定了硝酸鎵對alp轉錄物穩態水平的影響,則得到類似的結果。在鈦圓片不存在下添加鎵鹽,與沒有這一添加劑的試驗相比,對alp轉錄物水平的影響很小;而在ti-ca-聚p圓片存在下,如果與沒有該補充物的試驗(從0.030±0.003增加到0.074±0.007;還見圖8)相比,也觀察到對ca-聚p引起的alp轉錄物水平增加的強協同效應(從0.027±0.003增加到0.115±0.007)。在未包被的鈦合金圓片上培養的細胞,沒有觀察到可檢測的或僅觀察到非常小的轉錄物水平。
表1.鎵對編碼caix和alp的基因表達的影響。在試驗混合物中不存在或存在100μm硝酸鎵時,如圖8的圖例所述進行實驗。將表達值標準化為gapdh的表達。在沒有任何鈦圓片、或者在鈦合金圓片、或者在ti-ca-聚p圓片上培養細胞。首先將培養物在mac不存在下孵育3d,然后轉移到補充有mac的培養基中,繼續孵育另外3d或5d。nd,不可檢測。
xrd分析
通過應用粉末x射線衍射(xrd)法,進行“ha”以及聚p-ha顆粒的相鑒定(圖10)。而對于純na-聚p而言,沒有明確的衍射信號可以被解析,表明是非晶形相,純的“ha”以及“ha(2.5)聚p”和“ha(5)聚p”表現出寬的衍射峰,表明形成低結晶度的ha;沒有檢測到其他結晶相(jcpds[http://www.icdd.com/]#09-0432)。然而,當聚p的量增加至10wt.%時,如在“acap(10)聚p”中,在xrd圖案中沒有看到結晶的跡象(圖10)。這些結果表明,隨著聚p含量的增加,制備的ha樣品的結晶度逐漸降低。
fitr光譜分析
在這里記錄的cap的所有光譜[如純“ha”以及“ha(2.5)聚p”和“ha(5)聚p”],顯示了典型的ha條帶(圖11a和圖11b),除了“acap(10)聚p”。如預期的那樣,在1090cm-1,1015cm-1和960cm-1處記錄了具有高強度的ha的典型吸收帶,具有對稱v1(po43-)伸縮振動和不對稱v3(po43-)伸縮振動。此外,在556cm-1和604cm-1處的v4(po43-)峰是特征彎曲振動。相比之下,“acap(10)聚p”的光譜顯示了在1100-900cm-1的區域中對稱v1(po43-)伸縮振動和不對稱v3(po43-)伸縮振動以及出現為以610cm-1附近為中心的一個峰的p=o彎曲振動的v4(po43-)諧波的磷酸鹽吸收帶的明顯移動。另外,從~3600cm-1直至3100cm-1的范圍的寬吸收帶指向v3和v1,具有拉伸模式的與氫鍵合的h2o分子。在1629cm-1處的吸收帶歸因于h2o分子的變形模式v2,證明在合成樣品中物理吸附水的存在。據報道,cap的ftir光譜中556cm-1和604cm-1附近的振動帶,反映了結晶po43-的諧波振動的特征彎曲信號;兩個峰的移位表明從結晶相向非晶形相的轉變。在“acap(10)聚p”的圖中清楚地看出這種移動,其中兩個峰現在顯示為一個峰,表明該樣品的非晶形性質。這個發現也與報道的xrd圖案一致(圖10)。
為了比較,聚p的譜也包括在cap追蹤中(圖11)。顯然的,對于聚p,1261cm-1附近的峰顯示屬于體現聚p特征的(o-p=o)的不對稱拉伸模式。接近1090cm-1和960cm-1的吸收帶分別屬于(o-p-o)的不對稱拉伸模式和對稱拉伸模式。這些信號進一步證實了聚p的存在。此外,864cm-1附近的吸收帶指示了p-o-p連接的不對稱拉伸模式,和以763cm-1附近為中心的部分分裂帶應屬于這些連接的對稱拉伸模式。
tem和粒度分布結果
通過tem分析cap樣品的形態。“ha”樣品顯示平均長度為39±8nm和寬度為14±4nm的針狀納米晶體(圖12a)。在長度為42±10nm和寬度為9±5nm的“ha(2.5)聚p”樣品中,可以看出幾乎相同的尺寸(圖12b)。“ha(5)聚p”制品中存在的晶體稍長為56±12nm且寬度為6±3nm(圖12c)。相比之下,含有最高比例聚p的cap制品(“acap(10)聚p”)顯示出具有不同形態的顆粒(圖12d)。不同于解析的直徑為70nm至120nm(96±15nm)的針狀結構球形顆粒。那些顆粒具有聚集成較大實體的傾向。
cap樣品和ca-聚p納米顆粒對細胞生長的影響
通過應用mtt試驗來檢測在cap樣品上的saos-2細胞的細胞存活力和生長。將那些樣品以100μg/ml的濃度加入細胞。并行地,用10μg/ml的ca-聚p納米顆粒“aca-聚p-np”進行孵育,所述“aca-聚p-np”是經證明可增加細胞生長速率并導致alp和col-i基因表達增加的樣品。
結果表明,在2d的孵育期后,在不同基底上沒有看到細胞生長的顯著差異。然而,在3d的孵育期后,在“aca-聚p-np”存在下測量到saos-2細胞生長的顯著增加(從1.74±0.19吸光度單位[第2天]至2.45±0.20吸光度單位)。在不同cap樣品上生長的增加較低,對于“ha”(從1.54±0.18至1.93±0.21)和“acap(10)聚p”(從1.54±0.18至2.31±0.25)生長的增加是顯著的。
sem分析
將細胞在純“ha”上或在“acap(10)聚p”圓片上培養3d。然后將樣品用多聚甲醛固定,并通過sem檢查。可以看出,在兩種試驗中,對于“ha”和“acap(10)聚p”培養物,細胞均牢固地附著于基底。在較高放大倍數下,細胞擴散的特性變得明顯。
cap上的saos-2細胞的基因表達傾向
最初將骨相關saos-2細胞培養3d,然后轉移到新的缺少mac或補充mac并且還含有cap樣品(100μg/ml)或聚p納米顆粒(10μg/ml)培養基中。然后在qrt-pcr分析之前繼續孵育7d,以確定col-1或alp轉錄物的穩態水平(圖13)。
測定顯示,在接種細胞時col-1的表達低且為0.26±0.07個表達單位,其與gapdh的表達相關。在不存在mac和存在cap樣品或聚p納米顆粒7d的情況下,與“ha(2.5)聚p”(至0.35±0.05)、“ha(5)聚p”(至0.43±0.05)以及“acap(10)聚p”(至0.52±0.06)孵育之后,表達水平顯著增加,如預期的那樣,對于聚p“aca-聚p-np”(至0.63±0.07)也是一樣;圖13a。暴露于mac后,所有穩態表達水平顯著較高,并且例如“ha”的值達到0.41±0.05,和“acap(10)聚p”的值達到1.06±0.11。后者“acap(10)聚p”的值接近暴露于聚p納米顆粒的細胞中具有的1.38±0.16的誘導水平。
如果與參考基因gapdh相關,則記錄alp基因相當的誘導表達模式。再次,與在mac存在下孵育7d的細胞的測量值相比,在mac不存在下alp表達水平較低(圖13b)。只有在沒有mac但補充有聚p納米顆粒的試驗中,alp的表達水平的增加是顯著的(從0.12±0.02到0.17±0.01)。然而,暴露于mac后,所有含聚p的cap制品的表達水平顯著高于細胞接種期間所見到的表達水平。對于“ha(2.5)聚p”的增加值為0.15±0.03,對于“ha(5)聚p”的增加值為0.28±0.03,和對于“acap(10)聚p”的增加值為0.89±0.09。再次,如果與含有較少量聚p的樣品相比,后一種表達水平更接近于具有1.37±0.16的暴露于聚p的細胞所測定的值。
聚p對方解石形成的影響:ftir光譜和xrd光譜
對于所有的caco3固體,記錄以下ftir信號:在≈1080cm-1處的v1(對稱伸縮),在≈870cm-1處的v2(平面外彎曲),在≈1400cm-1處的v3(雙重退化平面不對稱伸縮)和在≈700cm-1處的v4(雙重退化平面彎曲)。用ftir/kbr小球獲得的發表的ir數據(rodriguez-biancojd,shaws和benninglg(2011)thekineticsandmechanismsofamorphouscalciumcarbonate(acc)crystallizationtocalcite,viavaterite.nanoscale3:265-271),包括方解石的位于約1400cm-1(v3)的峰、876cm-1(v2)的峰和714cm-1(v4)的峰,以及球霰石的位于1090cm-1(v1)的峰、870cm-1(v2)的峰和745cm-1(v4)的峰(圖14)。在聚p不存在下制備的我們的樣品的特征如下。對于方解石,記錄了典型的振動帶1391cm-1、872cm-1和712cm-1,而在聚p存在下制備的樣品,對于“ccp5”聚p在1398cm-1、869cm-1和742cm-1處顯示出吸附峰,以及對于“ccp10”顯示在1398cm-1、869cm-1和741cm-1處的帶,證明了球霰石的形成。顯然,在制備的caco3固體中(“ccp10”對“ccp5”),聚p含量較高時,對于球霰石在741cm-1附近的信號強度降低。這表明acc的形成。除co32-的吸收峰外,從1200cm-1到950cm-1的峰對應于聚p中磷酸鹽的吸收峰。
用xrd證實了上述結果,其中給出了在聚p不存在下制備的樣品在大約23°、30°、36°和40°處的衍射峰;那些信號對應于方解石。相反,在聚p(“ccp5”)存在下制備的樣品在大約24°、27°、32°和44°處顯示出峰,這也反映了球霰石的存在。此外,這些數據證明,在聚p不存在下制備的caco3固體是純方解石(圖15a),而“ccp5”樣品由與acc結合的球霰石組成,其可以從樣品“ccp5”的低強度信號以及衍射峰的加寬來推理(圖15b)。因此,如“ccp10”中聚p的量的增加,降低了acc向球霰石的轉化率。明顯地,來自“ccp10”樣品的xrd圖案,其顯示了樣品的非晶形性質,但是還含有少量的球霰石。
形成的固體形態
通過sem研究了由cacl2·2h2o和na2co3沉淀形成的固體。在聚p不存在下形成的顆粒其顯微照片顯示典型的結晶方解石,由{104}面包圍的菱面體晶體,圖16a和圖16b。顆粒的尺寸在5.3μm至8.9±2.4μm之間變化。相反,在聚p存在下由cacl2·2h2o和na2co3形成的固體顯示不同的形態。在較低的聚p濃度下,“ccp5”顆粒呈球形外觀,球形晶體的平均尺寸為9.4±3.7μm(圖16c和圖16d);我們將這些顆粒歸屬于球霰石。它們被非常豐富地積累的尺寸范圍為100nm至200nm的小納米顆粒包圍,我們將其指定為acc。如“ccp10”中增加聚p,球狀顆粒消失,并被五角形片狀/六邊形片狀的顆粒(5-10μm大小的球霰石)取代(圖16e和16f)。
caco3樣品對細胞生長/存活力的影響
通過應用mtt試驗(參見上文)測定暴露于caco3制品后saos-2細胞的細胞生長/存活力。將caco3樣品以50μg/ml的濃度加入到細胞中。并行地,進行缺乏任何caco3固體的對照試驗。結果表明,在對照試驗和三種caco3系列(“ccp5”、“ccp10”或方解石)中,細胞生長/存活力從時間0的0.70±0.11增加到2d后的大約1.1個吸收單位和3d后的2.35個單位,僅有不顯著的變化。
caco3固體在培養基中的穩定性
saos-2細胞以附著方式生長(pautkec,etal.(2004)characterizationofosteosarcomacelllinesmg-63,saos-2andu-2osincomparisontohumanosteoblasts.anticancerres24:3743-3748)。如果培養物暴露于方解石或“ccp5”固體,培養皿表面上的生長行為,在含有“ccp10”(圖17a和圖17b)或方解石(圖17c和圖17d)的試驗中是類似的。3d后,細胞生長幾乎達到融合。然而,值得注意的是,與在方解石試驗中觀察到的那些相比,在含有“ccp10”的試驗中,在這段時間后漂浮在培養基中的礦物顆粒的數量顯著降低。該觀察結果可作為“ccp10”顆粒在5d孵育期內經歷溶解的指示。這一發現由測定支持,其顯示在模擬體液(oyanea,kimhm,furuyat,kokubot,miyazakit,nakamurat(2003)preparationandassessmentofrevisedsimulatedbodyfluids.jbiomedmaterresa65:188-195)中的3d孵育期后,基于可沉淀的碳酸鹽測量(數據未顯示),方解石顆粒的量僅降低5-10%,而只有35%的“ccp10”顆粒可被回收。
從caco3顆粒釋放ca2+
在單獨的試驗中,將方解石或“ccp10”加入用1mtris-hcl(ph7.4)緩沖的1ml試驗中。雖然幾乎沒有從方解石樣品釋放ca2+,但在48hr后,已經從“ccp10”釋放了6.8±1.1μg/ml(反應混合物中總ca2+的68%);在整個192hr孵化期間,這一程度進一步增加(圖18)。
在saos-2細胞以及msc中alp的表達
在不存在和存在mac的情況下,測定caco3樣品對saos-2細胞以及msc的形態發生活性。使用saos-2細胞,確定在mac不存在下,alp與參考基因表達(gapdh)之間的表達比顯著地從0.31±0.9增加到≈0.6。在沒有mac的實驗組中,沒有測量到顯著差異,而不管在試驗中caco3樣品是不存在(對照)還是存在(圖19a)。然而,如果在mac激活的細胞中測定表達比(alp:gapdh),則測量到比值顯著增加至0.87±0.12(在對照中)、增加至1.74±0.23(“ccp5”)、或增加至1.86±0.29(“ccp10”)。相反,在用方解石的試驗中,沒有測量到細胞的響應(0.14±0.05)。
如果將msc用于實驗,則發現如果與參考gapdh基因表達相關的alp的類似表達模式。再次,在任何caco3固體不存在下以及在“ccp5”和“ccp10”兩者存在下的試驗中,在mac存在下看到顯著增加的表達比。在暴露于方解石的細胞中沒有測定到誘導作用(圖19b)
在saos-2細胞中bmp2的表達
通過qrt-pcr分析測定bmp2對“ccp10”和聚p(ca2+絡合物)響應的表達水平。將saos-2細胞在礦化培養基(mccoy's培養基/mac)中孵育長達7天。在實驗開始時,向培養物中加入“ccp10”(50μg/ml)、聚p(ca2+絡合物;5μg/ml;對應于磷酸鹽的50μm)或方解石(50μg/ml)。終止后,從培養物中提取rna并進行qrt-pcr。使用管家基因gapdh的表達作為參考。如圖20所示,加入“ccp10”或聚p(ca2+絡合物)3至7天后,bmp2的表達水平顯著增加。然而,與聚p(ca2+絡合物)相比,“ccp10”的bmbp2表達增加更快。在“ccp10”的3天孵育期后,已經達到bmb2基因表達的最高水平,而由聚p(ca2+絡合物)誘導的該基因的表達僅在更長時間(5天孵育期)后達到最高水平(與“ccp10”相比類似的水平)。在第3天,與聚p(ca2+絡合物)相比,響應“ccp10”的bmp2的表達水平顯著高(約2倍),表明兩種組分的“協同”效應。7天后,“ccp10”和聚p(ca2+絡合物)的表達水平均降低,但依然保持顯著。在聚p不存在下,由亞穩態acc形成的方解石對bmp2基因表達沒有顯示任何刺激作用。
用于動物研究的微球
對照球“contmic”具有(≈845μm[820±60μm];n=50)的大小,而含有聚p的那些大小稍微小一些(≈838μm[816±65μm]);圖21a和圖21b。微球表面的結構是多孔的,具有25nm-30nm的孔(此處未示出)。“聚p-mic”中的聚p含量為7.26±0.92%。測定“contmic”和“聚p-mic”的顆粒硬度;“contmic”的中值redym剛度為26.99±6.22kpa,“聚p-mic”微球的中值redym剛度為23.96±23.96kpa。
大鼠中的相容性研究
如“材料和方法”所述,將微球樣品(20mg),“contmic”和“聚p-mic”嵌入大鼠背部的肌肉中。在2周、4周或8周后,取出帶微球的組織樣品,切片并用蘇木精溶液染色。在用于“contmic”和“聚p-mic”系列的每組所有三只處死的實驗室動物中,沒有在一個離體的標本中可以看到任何組織病理學改變的跡象。2周后,在微球已經放入肌肉的區域,一些細胞分散在微球區域內。然而,“cont-mic”微球在肌肉區域中停留4周和8周后,它們顯示是空的或接近無細胞的。相比之下,在“聚p-mic”微球內4周后,細胞在球體內的積累明顯。8周后,球體幾乎充滿了浸潤細胞。
8周后植入區域硬度的測定顯示了明顯增加的中值redym剛度,對于“cont-mic”是33.13±7.97kpa,對于“聚p-mic”微球是60.11±12.13kpa。在植入前,大鼠背部肌肉具有74.40±14.33kpa的中值redym剛度。
方法
聚磷酸鹽
已從chemischefabrikbudenheim(budenheim;德國)獲得實施例中使用的聚磷酸鈉(平均鏈為40個磷酸鹽單元的na-聚p)。
ti-ca-聚p圓片
鈦合金(ti-6a1-4v)圓片(直徑15mm和厚度2mm),可以從例如nobelbiocare獲得。使用前,用砂紙(碳化硅,matador)拋光,然后在蒸餾水中用超聲波清洗,隨后在丙酮(10min)和40%乙醇溶液(15min)中洗滌,最后在蒸餾水中漂洗20min。將樣品在50℃下干燥24小時。然后在室溫下將鈦合金圓片在20ml的5mhcl中孵育6h。在蒸餾水中溫和洗滌之后,將圓片在室溫下干燥,并在硅烷偶聯劑(3-氨丙基)三甲氧基硅烷[aptms](例如,來自sigma-aldrich)存在下,將處理的圓片樣品用10mlca-聚p納米顆粒懸浮液覆蓋。
通過將0.5gna-聚p與atpms溶液(1wt%)在100ml水中混合,然后加入0.1gca2+氯二水合物(cacl2·2h2o)來制備ca-聚p微粒。將鈦圓片于90℃在上述懸浮液中孵育3小時;在這些條件下,最初形成膠體懸浮液。將環境ph調節至8.0,以便通過ca2+離子鍵/橋接將聚p結合到硅烷蝕刻的鈦圓片。樣品保留在該懸浮液中1d。研究了兩種不同的atmps濃度(分別為1mg/試驗和2mg/試驗)對在鈦表面上形成的包被層形態的影響。最后,取出樣本鈦-鈣-聚p(ti-ca-聚p)圓片,并在100℃干燥(參見圖1)。
在實施例中描述的實驗中,如果沒有提及其他的,則使用以較高比例的aptms然后與ca-聚p一起制備的圓片。
ha和聚p-羥基磷灰石的合成
可以通過濕法化學沉淀法,由氯化鈣(cacl2)作為ca2+源和磷酸氫二銨((nh4)2hpo4)作為磷酸鹽源合成羥基磷灰石(ha)納米粒子。為了沉淀化學計量的ha(ca10(po4)6(oh)2;ca/p比為1.667),將100ml0.3m(nh4)2hpo4水溶液逐滴加到100ml0.5mcacl2水溶液中。為了獲得ca/p摩爾比為10:6的ha,計算試劑的量。加入氫氧化鈉溶液,使反應的ph值保持在10。
為了制備各種聚p含量的聚p取代的ha納米顆粒,起始成分(cacl2和(nh4)2hpo4)還如下補充有2.5wt.%、5wt.%或10wt.%的na-聚p(稱為ha,或各自的cap制品)。將各自量的na-聚p、0.12g[“ha(2.5)聚p”]、0.25g[“ha(5)聚p”]或0.50g[“acap(10)聚p”]加入到(nh4)2hpo4水溶液;然后將該溶液加入到溶解的cacl2中。將ph保持在10。將所得沉淀物在室溫下放置24h。然后過濾沉淀,用蒸餾水洗滌3次,然后在60℃的熱風烘箱中干燥24h。最終粉末稱為“ha”、“ha(2.5)聚p”、“ha(5)聚p”和“acap(10)聚p”。
制備聚p納米顆粒
對于比較功能/生物學研究,可以如所述制備非晶形ca-聚p納米顆粒(müllerweg,tolbae,
制備碳酸鈣微粒
使用等摩爾濃度比ca2+和co32-的cacl2·2h2o溶液和na2co3溶液,通過快速混合在水溶液(室溫)中直接沉淀制備碳酸鈣(caco3),示意圖見圖3。
為了研究聚p對沉淀的caco3的影響,向其中補充有0.05g或0.1gna-聚p的20ml0.1mnaoh的溶液中加入1.05gna2co3;隨后將該溶液用30ml去離子水稀釋。然后加入含有1.47gcacl2·2h2o的50ml水。由此,分別向caco3沉淀試驗中加入5%[w/w](加入0.05gna-聚p)和10%[w/w](0.1gna-聚p)聚p。將獲得的懸浮液過濾,用丙酮洗滌并在室溫下干燥。樣品稱為“ccp5”(每次caco3沉淀試驗為0.05gna-聚p)或“ccp10”(0.1g)。
ca-聚p包被層的耐久性
可以例如通過測定來自圓片的ca2+釋放,來量化鈦圓片周圍的ca-聚p包被層的穩定性和耐久性。將對照圓片以及ti-ca-聚p盤浸沒在模擬體液(sbf)中,但是省略ca2+作為組分;將ph調節至8.0。試驗體積為1ml,并且在37℃下進行孵育。通過應用絡合滴定法測定ca2+濃度;使用試劑eriochromeblackt(例如,來自sigma-aldrich)。在實施例中描述的實驗中,在圓片一個平面上的聚p包被層的表面厚度,已通過顯微鏡確定為≈5μm。反過來,在圓片一個平面上的ca-聚p(密度≈3g/ml)的總量,具有≈2.4mg的值。
如實施例所示,將5μg來自牛腸粘膜的堿性磷酸酶(alp)(例如來自sigma;≥6,500dea單位/mg蛋白)加入到反應混合物中。
顯微鏡分析
可以例如用來自keyence的、配備有vh-z25變焦鏡頭(25x至175x放大)或vh-z-100長距離高性能變焦鏡頭(高達1000x放大)的vhx-600數碼顯微鏡,來進行圓片的光學顯微鏡檢查。可以例如通過使用keyencevk分析儀軟件,來測量表面粗糙度。對于掃描電子顯微鏡(sem)分析,可以使用例如hitachisu8000電子顯微鏡(hitachihigh-technologieseuropegmbh,krefeld,德國)。
電子顯微鏡和能量色散x射線光譜
對于透射電子顯微鏡(tem)分析,例如可以使用在120kv的加速電壓下在tecnai12透射電子顯微鏡(fei)上操作的temcam-f416(4k×4k)ccd照相機(tvips)。該設備與粒度分析儀(imagej)連接;在實施例中描述的實驗中,已經評估了25-50個晶體/球體。
可以例如在低電壓(1kv)用su8000儀器(hitachihigh-technologieseurope),進行掃描電子顯微鏡(sem)分析。對于實施例中所述的研究,使細胞于6孔板中在已經壓制成直徑為34mm、厚1mm圓片的cap制品上生長3天。用4%多聚甲醛固定在cap基底上生長的細胞。
可以例如用連接到掃描電子顯微鏡(nova600nanolab;fei),在10kv操作且收集時間為30-45s的edaxgenesisedx系統,進行能量色散x射線(edx)光譜。分析大約5μm2的區域。
可以用例如hitachisu8000顯微鏡,在低電壓(<1kv,近表面有機表面的分析)下進行edx映射。將sem與xflash5010檢測器(允許同時進行基于edx的元素分析的x射線檢測器)相結合。設備的這種組合用于更高電壓(10kv)分析,在此期間將xflash5010檢測器用于沉積物表面的元素映射。hypermap數據庫用于解釋。
x射線衍射分析
可以如所述進行x射線衍射(xrd)實驗(raynauds,etal.calciumphosphateapatiteswithvariableca/patomicratioi.synthesis,characterizationandthermalstabilityofpowder.biomaterials2002;23:1065-1072)。可以例如在具有cukα輻射(
傅里葉變換紅外光譜
可以通過使用微磨(瑪瑙研缽和杵)礦物粉末,例如在配備有goldengateatr部件(specac)的atr-ftir分光鏡/varian660-ir光譜儀(agilent)中,進行傅立葉變換紅外(ftir)光譜分析。實施例中所示的每個光譜代表具有4cm-1(通常為550-1800cm-1)的光譜分辨率的100次掃描的平均值。可以例如用varian660-ir軟件包5.2.0(agilent),實現光譜的基線校正、平滑化和分析。可以例如用originpro(版本8.5.1;originlab)進行光譜的圖形顯示和注釋。
從caco3顆粒釋放ca2+
在單獨的試驗中,將100μg/ml方解石或“ccp10”加入到含有1ml1mtris-hcl(ph7.4)的eppendorf管中。在室溫下孵育2h、2d、3d和8d后,取出100μl的樣品并離心,分析上清液中ca2+濃度。可以例如用光度測試試劑盒(例如,millipore/merckchemicals;貨號100858“calciumcelltest”)進行測定。從測試試驗中減去空白值。
培養saos-2細胞
將骨細胞如saos-2細胞(人類成骨肉瘤細胞),在補充有2mml-谷氨酰胺、10%或15%熱滅活胎牛血清(fcs)和100單位/ml青霉素和100μg/ml鏈霉素的mccoy培養基(biochrom-seromed)中培養。將細胞在25-cm2培養瓶或6孔板(表面積9.46cm2;例如來自orangescientifique)中,在37℃的濕潤培養箱中孵育。常規地,以3×104個或1×104個細胞/孔開始培養,總體積為3ml。在此指出,培養物首先在不存在礦化-激活混合物(mac)中,在包含5mmβ-甘油磷酸酯、50mm抗壞血酸和10nm地塞米松的情況下培養3d。然后在不存在或存在mac的情況下,將培養物繼續孵育多達7d。在實驗開始時,向每個孔中加入ha/聚p礦物樣品(100μg/ml[ha,cap]或10μg/ml[“aca-聚p-np”])。每三天,用新鮮的培養基/血清替換培養基,并且在此指出也有mac。對于使用圓片的研究,使用24孔板(例如,來自corning;每個孔的直徑為15.6mm),其中插入15mm大的圓片。以2ml細胞/培養基/fcs的總體積進行試驗。
在實施例中所示的另一系列實驗中,在100μm硝酸鎵存在下進行了試驗。
細胞增殖/細胞存活力試驗
可以例如通過基于四唑鹽xtt(例如,細胞增殖試劑盒ii(roche))或3-[4,5-二甲基噻唑-2-基]-2,5-二苯基四唑(mtt;#m2128,sigma)的比色測定法,來測定細胞增殖/生長(wangxh,etal.(2014)modulationoftheinitialmineralizationprocessofsaos-2cellsbycarbonicanhydraseactivatorsandpolyphosphate.calciftissueint94:495-509)。
人類間充質干細胞
用人間充質干細胞(msc)測定alp的表達,在saos-2細胞平行測定alp的表達。使用已建立的方法分離和培養細胞(wangxh,etal.themarinesponge-derivedinorganicpolymers,biosilicaandpolyphosphate,asmorphogeneticallyactivematrices/scaffoldsfordifferentiationofhumanmultipotentstromalcells:potentialapplicationin3dprintinganddistractionosteogenesis.marinedrugs12,1131-1147)。
逆轉錄定量實時pcr分析
應用定量實時rt[逆轉錄]-pcr(qrt-pcr)技術,來確定圓片對saos-2細胞中以下基因表達水平的影響。簡言之,從細胞中提取rna,使用以下引物對進行pcr反應:碳酸酐酶ix(caix;nm_001216人)fwd:5'-acatatctgcactcctgccctc-3'[nt977至nt998](seqidno1)和rev:5'-gcttagcactcagcatcactgtc-3'[nt1105至nt1083](seqidno.2),堿性磷酸酶(alp;nm_000478.4)fwd:5'-tgcagtacgagctgaacaggaaca-3'[nt1141至nt1164](seqidno.3)和rev:5'-tccaccaaatgtgaagacgtggga-3'[nt1418至nt1395](seqidno.4),i型膠原蛋白(coli;nm_000088.3)fwd:5'-gactgccaaagaagccttgcc-3'[nt5073至nt5093](seqidno:5)和rev:5'-ttcctgactctcctccgaaccc-3'[nt51196至nt5175](seqidno:6)和bmp2(骨形態發生蛋白2;nm_001200.2)fwd:5'-accctttgtacgtggacttc-3'[nt1681至nt1700](seqidno:7)和rev:5'-gtggagttcagatgatcagc-3'[nt1785至nt1804](seqidno:no:8)。使用甘油醛-3-磷酸脫氫酶(gapdh)作為參考(nm_002046.5)fwd:5'-ccgtctagaaaaacctgcc-3'[nt929至nt947](seqidno.9)和rev:5'-gccaaattcgttgtcatacc-3'[nt1145至nt1126](seqidno.10)。可以例如在icycler(bio-rad)中,應用各自的icycler軟件進行pcr反應。在確定ct值后,計算各個轉錄物的表達。
制備基于plga的微球
詳細描述制備用于動物實驗的微球體(wangsf,etal.(2014)bioactiveandbiodegradablesilicabiomaterialforboneregeneration.bone:67:292-304)。使用4mlplga/二氯甲烷溶液(體積比1:5)制備缺少ccp10的微球體;它們被稱為“cont-mic”(plga:聚(d,l-丙交酯-共-乙交酯);丙交酯:乙交酯[75:25];mol.wt.66,000-107,000)。對于制備含有caco3/聚p的微球體,以20%的濃度將“ccp10”微球體(“聚p-mic”)加入到plga/二氯甲烷混合物中。通過具有0.8mm孔徑的注射器壓制粘性反應混合物。通過這種方法,獲得平均直徑≈820μm的微球體。
如所述測定微球體中聚p的含量(mahadevaiahms,etal.(2007)asimplespectrophotometricdeterminationofphosphateinsugarcanejuices,wateranddetergentsamples.e-journalofchemistry4:467-473)。
測定機械特性
可以例如用配備有已經被熔合到套圈光纖頂部的懸臂的納米壓痕儀,來測定微球體和植入區域(再生區域)的肌肉組織的機械特性(wangsf等人(2014)bioactiveandbiodegradablesilicabiosaterialforboneregeneration.bone67:292-304)。使用該技術來量化降低的楊氏模量(redym)。
體內相容性研究
在實施例中描述的實驗中,使用重量在240g和290g(年齡:兩個月)之間的(雄性)性別的wistar大鼠;每組使用3只動物。在整個實驗期間,隨意提供飲食和飲水。在手術前,用10ml/kg體重的環丙沙星處理動物,用于抗生素預防。然后通過肌內注射用氯丙嗪/氯胺酮對動物進行麻醉。接下來在垂直于上肢水平的椎軸的右側半部和左側半部,進行≈1cm切口的常規消毒。皮膚切開后,將肌肉切開并分開以容納微球。將微球體(100μl體積,≈20mg)引入肌肉中,并在更深的層中穩定(vidyas.,parameswarana.,sugumaranvg(1994)comparativeevaluationoftissue.compatibilityofthreerootcanal.sealantsinrattusnorwegicus:ahistopathologicalstudy.endodontology6:7-17)。經過2周、4周或8周的時間,處死動物,切開有周圍組織的樣本并切片。對樣品宏觀地檢查炎癥、感染和變色。將樣品固定在福爾馬林中,切片,用梅氏蘇木精染色,然后通過光學顯微鏡(例如,用olympusahbt3顯微鏡)進行分析。
統計分析
使用配對studentt檢驗對結果進行統計學評估。
序列表
<110>維爾納·恩斯特·路德維格·格奧爾格·米勒
<120>作為具有形態發生活性的包被層和支架的非晶形無機聚磷酸鹽-磷酸鈣和碳酸鈣顆粒
<130>m32732wo02
<150>gb1420363.2
<151>2014-11-17
<150>gb1513011.5
<151>2015-07-23
<150>gb1515515.3
<151>2015-09-02
<150>gb1518575.4
<151>2015-10-20
<160>10
<170>patentinversion3.5
<210>1
<211>22
<212>dna
<213>智人(homosapiens)
<400>1
acatatctgcactcctgccctc22
<210>2
<211>23
<212>dna
<213>智人
<400>2
gcttagcactcagcatcactgtc23
<210>3
<211>24
<212>dna
<213>智人
<400>3
tgcagtacgagctgaacaggaaca24
<210>4
<211>24
<212>dna
<213>智人
<400>4
tccaccaaatgtgaagacgtggga24
<210>5
<211>21
<212>dna
<213>智人
<400>5
gactgccaaagaagccttgcc21
<210>6
<211>22
<212>dna
<213>智人
<400>6
ttcctgactctcctccgaaccc22
<210>7
<211>20
<212>dna
<213>智人
<400>7
accctttgtacgtggacttc20
<210>8
<211>20
<212>dna
<213>智人
<400>8
gtggagttcagatgatcagc20
<210>9
<211>19
<212>dna
<213>智人
<400>9
ccgtctagaaaaacctgcc19
<210>10
<211>20
<212>dna
<213>智人
<400>10
gccaaattcgttgtcatacc20
- 還沒有人留言評論。精彩留言會獲得點贊!