一種納米纖維素/聚多巴胺復合智能凝膠藥物緩釋材料的制備方法與流程
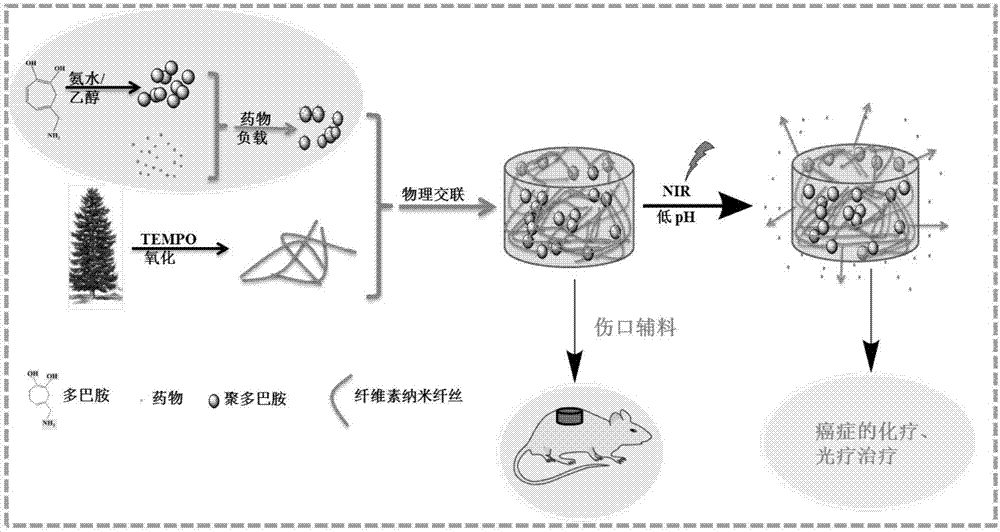
本發明屬于醫用材料技術領域,具體涉及納米纖維素/聚多巴胺復合凝膠藥物緩釋材料的制備方法。
背景技術:
藥物緩釋是將小分子藥物與高分子材料相結合,利用高分子材料固有的相關特性(如ph敏感性和溫度敏感性等)使藥物分子在高分子載體中緩慢釋放出來。藥物緩釋體系與傳統給藥體系相比較,其能夠有效地提高藥物的生物利用率,減少給藥次數以及提高用藥的穩定性。因此制備藥物緩釋材料引起研究人員的廣泛興趣。如江蘇天竹化工科技有限公司研制開發的可用于藥物緩釋方向的殼聚糖丙烯酸酯類藥物復合材料,美國3m公司研制開發的封裝并實現藥物控制釋放的多層結構復合材料,天津凱文生物科技有限公司研制開發的曲美他嗪緩釋膠囊等。
水凝膠是指親水性聚合物通過化學或者物理作用形成的三維交聯的網絡結構,其不溶于水但能吸收大量的水,具有良好的溶脹性能、力學性能、生物相容性等特性,在藥物緩釋和皮膚創傷敷料中得到了廣泛的應用。在文獻(macromolecules,2000,33(1),102-107)中zhang等人報道了n-異丙基丙烯酰胺水凝膠的低臨界溶解溫度(lcst)為33℃,其非常接近人體的溫度,故成為了目前最廣泛研究的溫敏性水凝膠。此外,ph敏感型水凝膠(如丙烯酸酯類)能根據周圍環境ph的改變而實現凝膠網絡的收縮和舒展,進而實現對所載藥物的ph響應釋放。然而,傳統水凝膠藥物緩釋體系存在很多缺點:由于水凝膠材料一般為親水均相的體系,其所負載藥物的釋放曲線大多數為非線性的釋放。此外,藥物釋放初期可能存在藥物突然釋放的現象,而釋放后期藥物濃度往往較低,無法達到治療的效果。將天然高分子材料(淀粉、纖維素、殼聚糖等)引入智能水凝膠改變其藥物緩釋特性及提高其生物相容性引起了科研工作者們廣泛的關注。但是,到目前為止,關于納米纖維素型的高效藥物緩釋水凝膠載體及其應用的報道十分有限。
此外,腫瘤疾病是困擾人類的最大難題,化療已經成為當前實體瘤化療的研究方向和熱點(見專利zl200410035923.7和專利us5651986)。但是,目前可生物降解的緩控釋制劑多為固態聚合物如聚乙醇酸或聚乳酸等為緩釋載體。由于此類高分子載體具有較高的疏水特性,這些聚合物在制備緩釋制劑的過程中需要采用有機溶劑(如二氯甲烷、二甲基甲酰胺、氯仿等)。為除去有毒的有機溶劑,必須進行干燥處理。研究發現,有機溶劑或者高熱過程常導致許多抗癌有效成分降解變性。
技術實現要素:
針對上述現有技術的問題,本發明的目的是提供一種納米纖維素/聚多巴胺復合凝膠藥物緩釋材料及其制備方法,該藥物緩釋材料可以應用于多種藥物緩釋領域,特別適合傷口治療和腫瘤術后治療等方面。
為實現上述目的,根據本發明的一個方面,提供了一種納米纖維素/聚多巴胺復合凝膠藥物緩釋材料包括:納米纖維素、聚多巴胺和負載在聚多巴胺上的藥物,其中基于100重量份的納米纖維素,所述聚多巴胺的量為0.1重量份至2重量份;基于100重量份的聚多巴胺,所述藥物負載量為1重量份至15重量份。
優選地,所述聚多巴胺的量為0.1重量份至0.8重量份;所述藥物負載量為1重量份至14重量份。
根據本發明的另一個方面,提供了一種所述納米纖維素/聚多巴胺復合凝膠藥物緩釋材料的制備方法,包括以下步驟:
(1)將聚多巴胺和藥物均勻混合,磁力攪拌一段時間,離心,將上層未負載上的藥物倒出,沉淀下來的成分用去離子水反復洗滌幾次,得到負載藥物的聚多巴胺溶液;
(2)將一定濃度的納米纖維素溶液和負載一定藥物量的聚多巴胺溶液混合均勻,倒入成型器中,緩慢滴加作為交聯劑的鈣離子溶液,靜止一段時間,可以得到納米纖維素/聚多巴胺復合智能凝膠藥物緩釋材料。
優先地,步驟(1)中,所述聚多巴胺根據現有文獻(scientificreports,2014,4,6070)中所記載的方法制備。
優先地,步驟(1)中,所述負載的藥物可以是疏水性藥物或親水性藥物,優選地選自疏水性或親水性抗腫瘤藥物和疏水性或親水性抗生素類藥物,進一步優選地選自紫杉醇、阿霉素、吡喃阿霉素、鹽酸四環素、青霉素和鹽酸阿霉素等。
優先地,步驟1)中所述聚多巴胺負載藥物的時間為5h-120h,基于100重量份的聚多巴胺,所述藥物負載量為1重量份至15重量份,優選為1重量份至14重量份。
優先地,步驟(2)中,所述納米纖維素是經過傳統tempo氧化法、羧甲基化、高碘酸鹽氧化、過硫酸鹽氧化等方法得到,其納米纖維素的質量濃度范圍至少大于1%且小于10%。
優先地,步驟(2)中,所述作為交聯劑的鈣離子可以選自氯化鈣、葡萄糖酸鈣、磷酸二氫鈣、硝酸鈣、碳酸氫鈣、硫酸氫鈣、亞硫酸氫鈣、次氯酸鈣、溴化鈣、碘化鈣、氯酸鈣、高氯酸鈣、高錳酸鈣等可溶性鈣鹽中的一種或多種,鈣離子的質量濃度為0.1%-10%。
優先地,步驟(2)中,所述制備緩釋材料所需的靜止時間為2h-48h。
根據本發明的另一個發明,提供了一種納米纖維素/聚多巴胺復合凝膠藥物緩釋材料的用途,所述用途包括應用于傷口治療和癌癥手術的后期治療。
優先地,當負載的藥物為抗生素類藥物(鹽酸四環素、青霉素等)時,該緩釋材料可以應用于傷口愈合。
優先地,當負載的藥物為抗腫瘤藥物(紫杉醇、阿霉素、吡喃阿霉素等)時,該緩釋材料可以應用于腫瘤手術的后期治療。
有益效果
1、本發明添加的聚多巴胺作為負載藥物載體,利用聚多巴胺的高粘附特性,對親水性和疏水性藥物都可以進行負載,同時增加了藥物負載量,克服一般材料只能負載親水性藥物的局限性。
2、該智能復合凝膠材料制備采用物理交聯,制備比較簡單,比較溫和,能夠節約成本。
3、納米纖維素/聚多巴胺復合凝膠藥物緩釋材料經過交聯液交聯后,具有一定的凝膠性能,能夠吸附血液中的水分,致使血液濃度增大,增加復合凝膠材料的止血性能,有一定的傷口愈合效果。
4、納米纖維素/聚多巴胺復合智能凝膠藥物緩釋材料中的聚多巴胺本身具有一定的抗菌性能,能夠有效提升材料的抗菌性能。
5、納米纖維素/聚多巴胺復合智能凝膠藥物緩釋材料具有一定的ph和近紅外光敏感性,可以應用于腫瘤手術的后期治療。
6、納米纖維素和聚多巴胺均具有生物相容性,可完全降解,易于廢棄處理,有利于保護環境,而且納米纖維素是天然可再生的綠色材料。
附圖說明
圖1為根據本發明制備所述納米纖維素/聚多巴胺復合凝膠藥物緩釋材料的工藝流程圖。
圖2為實施例1所得的復合水凝膠的掃描電鏡分析圖。
圖3為實施例1、實施例2、實施例3、實施例4中隨著負載時間的增加,聚多巴胺負載藥物量不斷增加,其中12h為實例1中對應藥物負載量,24h為實例2中對應藥物負載量,36h為實例3中對應藥物負載量,48h為實例4中藥物負載量。
圖4為本實施例2中所得的復合水凝膠載體進行近紅外光照射,其藥物緩釋規律圖。
圖5為實施例3中所得的復合水凝膠載體在不同的ph緩沖液體系中進行長時間的藥物緩釋圖。
圖6為實施例1中所得的復合水凝膠載體的抗菌效果圖,其中a:納米纖維素水凝膠;b:納米纖維素/聚多巴胺水凝膠;c:納米纖維素/聚多巴胺/鹽酸四環素水凝膠。
圖7為實施例5中采用的聚多巴胺的掃描電子顯微鏡(sem)和透射電子顯微鏡(tem)照片,其中a、b為sem照片;c、d為tem照片。
具體實施方式
在下文的描述之前,應當了解在說明書和所附權利要求中使用的術語,并不應解釋為局限于一般及辭典意義,而是應當基于允許發明人為最好的解釋而適當定義術語的原則,基于對應于本發明技術層面的意義及概念進行解釋。因此,在此的描述僅為說明目的的優選實例,而并非是意指限制本發明的范圍,因而應當了解的是,在不偏離本發明的精神和范圍下可以做出其他等同實施和修改。
根據現有技術的問題,本發明的發明人研發了一種新型的納米纖維素基藥物緩釋凝膠材料。本發明具有制備工藝簡單、反應溫和的特點。通過本發明方法制備得到的納米纖維素/聚多巴胺復合智能凝膠藥物緩釋材料具有良好的生物相容性和可降解性,并能有效促進傷口中血小板的聚集,有利于傷口愈合,同時該凝膠具有一定的近紅外光和ph敏感性,此外對細胞毒副作用小,能廣泛的應用于傷口治療和癌癥的術后治療。
在根據本發明的所述納米纖維素/聚多巴胺復合凝膠藥物緩釋材料中,聚多巴胺主要起負載藥物的作用,鈣離子起交聯作用,納米纖維素主要起成凝膠和賦予該復合凝膠材料一定強度的作用。藥物緩釋的主要機理是,利用聚多巴胺的ph響應性和光熱效應,在指定的環境條件下(例如,特定的ph或近紅外光照射條件下),將藥物可控釋放出來,并通過該復合凝膠的網絡結構達到良好的藥物緩釋效果。
根據本發明的所述納米纖維素/聚多巴胺復合凝膠藥物緩釋材料具有一定的ph敏感性,可以隨著ph的降低,藥物釋放量逐漸增加。同時該材料還具有一定的近紅外光敏感性,隨著照射時間和照射功率的增加,藥物釋放量會增加,可應用于癌癥手術后的光療和化療。
在根據本發明的所述納米纖維素/聚多巴胺復合凝膠藥物緩釋材料中,優先地,基于100重量份的聚多巴胺,所述藥物負載量為1重量份至15重量份。當藥物負載量低于1重量份時,藥物緩釋不明顯;當藥物負載量高于15重量份時,藥物緩釋會出現明顯的突釋現象。
在根據本發明的所述納米纖維素/聚多巴胺復合凝膠藥物緩釋材料中,優先地,基于100重量份的納米纖維素,所述聚多巴胺的量為0.1重量份至2重量份。當聚多巴胺加入量小于0.1重量份時,藥物負載量過低,藥物緩釋效果較差;當聚多巴胺加入量大于2重量份時,會顯著降低復合凝膠的強度,不能滿足實際應用的需求。
在根據本發明的所述納米纖維素/聚多巴胺復合凝膠藥物緩釋材料的制備過程中,優先地,步驟1)中所述聚多巴胺負載藥物的時間為5h-120h,基于100重量份的聚多巴胺,所述藥物負載量為1重量份至15重量份。負載藥物的時間低于5h時,聚多巴胺上藥物負載量過低,藥物緩釋不明顯;負載藥物時間超過120h時,聚多巴胺的藥物負載量也會降低,這與攪拌時間太久,聚多巴胺結構受損有關。
優先地,步驟(2)中,所述納米纖維素是經過傳統tempo氧化法、羧甲基化、高碘酸鹽氧化、過硫酸鹽氧化等方法得到,其納米纖維素的質量濃度范圍至少大于1%。當納米纖維素的濃度低于1%時,其成凝膠效果較差;如果凝膠材料中不加入納米纖維素,聚多巴胺不能起到可控緩釋的作用。
本發明,利用納米纖維素獨特的結構性質(如高比表面積、高長徑比、低密度、優越的機械性能等),以其為藥物輔料,采用聚多巴胺為藥物載體,制備納米纖維素基智能藥物緩釋凝膠材料。聚多巴胺(polydopamine,pda)是海洋貽貝類生物體分泌蛋白質的主要成分,具有較好的生物相容性、殺菌性能。在chemicalreviews,2014,114(9),5057-5115中liu等人報道,pda結構中存在大量的酚羥基、氨基和亞氨基等功能基團,可以螯合過渡金屬離子尤其是fe3+,因此可以與血紅蛋白中的fe3+特異性結合,實現止血抗菌的效果。此外,由于聚多巴胺存在較高的粘附性,可以負載親水性和疏水性藥物,能夠解決藥物載體難以負載疏水性藥物的難題。
以下實施例僅是作為本發明的實施方案的例子列舉,并不對本發明構成任何限制,本領域技術人員可以理解在不偏離本發明的實質和構思的范圍內的修改均落入本發明的保護范圍。
實施例1
(1)將1mg/ml聚多巴胺10ml和5mg鹽酸四環素均勻混合,攪拌12h后,離心,將上層未負載上的藥物倒出,沉淀下來的成分用去離子水反復洗滌幾次,得到負載藥物的聚多巴胺分散液;
(2)將質量百分比濃度為1%納米纖維素和上述負載一定藥物量的聚多巴胺分散液混合均勻,聚多巴胺的加入量為0.2wt%(相對于納米纖維素的質量),倒入成型器中,緩慢滴加質量百分比濃度為5%的氯化鈣溶液(2ml),靜止24h,可以得到納米纖維素/聚多巴胺復合凝膠藥物緩釋材料。
檢測方法
(1)將所得到的藥物水凝膠制劑置于模擬人體的生理環境的pbs緩沖液中(ph=5和ph=7.4),水平震蕩(100r/min),每隔1小時取2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。緩沖液要現配現用。通過紫外分光光度計法測定介質中藥物的濃度。pbs(ph=7.4)緩沖液的配置:將磷酸二氫鉀0.24g、磷酸氫二鈉1.44g、氯化鈉8.0g、氯化鉀0.2g溶解到1l的容量瓶中得到;pbs(ph=5)緩沖液可以通過調節pbs(ph=7.4)得到。
(2)將步驟(2)中制備的智能復合凝膠藥物緩釋材料置于模擬人體的pbs緩沖液生理環境中(ph=5和ph=7.4),采用3w的近紅外光激光發射器進行照射,每隔10min照射一次,取出2ml溶液,并取相同量的緩沖液放回其中以維持總的溶液體積不變。通過紫外分光光度法測定介質中藥物的濃度。
(3)將制備的智能復合凝膠藥物緩釋材料先用醫用無水乙醇進行消毒處理,采用大腸桿菌為菌體,研究其抗菌性能。其抗菌效果如圖6所示,證明該凝膠材料具有一定的抗菌性。
對本實施例所得到的智能復合水凝膠載體進行掃描電鏡分析,所得的干燥水凝膠的sem圖如圖2所示。由圖可以看出,水凝膠是由納米纖維交錯形成的三孔網絡結構,聚多巴胺納米微粒子分散在凝膠結構中,起到一定的支撐作用。
根據下式計算藥物負載率:
藥物負載率(%)=wt/w0×100%(wt為t小時后pda上藥物負載量g,w0為總的藥物量g)。
根據下式計算藥物緩釋率:
藥物緩釋率(%)=wt/w0×100%(wt為t小時后水凝膠中釋放藥物質量g,w0為水凝膠中總的藥物包載量g)。
經過分析計算,本實例聚多巴胺藥物負載量為12%,如圖(3)所示。在ph為5的緩沖液體系中,該凝膠材料達到最大緩釋率(80%)的時間約為36h;在ph為7.4的緩沖液體系中,該凝膠材料達到最大緩釋率(60%)的時間約為35h,均具有良好的藥物緩釋持續性。
實施例2
(1)將1mg/ml聚多巴胺10ml和5mg鹽酸四環素均勻混合,攪拌24h后,離心,將上層未負載上的藥物倒出,沉淀下來的成分用去離子水反復洗滌幾次,得到負載藥物的聚多巴胺分散液;
(2)將質量百分比濃度為2%納米纖維素和上述負載一定藥物量的聚多巴胺分散液混合均勻,聚多巴胺的加入量為0.2wt%(相對于納米纖維素的質量),倒入成型器中,緩慢滴加質量百分比濃度為4%的葡萄糖酸鈣溶液(2ml),靜止24h,可以得到納米纖維素/聚多巴胺復合智能凝膠藥物緩釋材料。
檢測方法
(1)將所得到的藥物水凝膠制劑置于模擬人體的生理環境pbs緩沖液中(ph=5和ph=7.4),水平震蕩(50r/min),每隔1小時取2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。緩沖液要現配現用。按照實施例1中相同的計算方式通過紫外分光光度計法測定介質中藥物的濃度。
(2)將制備的復合凝膠藥物緩釋材料置于模擬人體生理環境pbs緩沖液中(ph=5和ph=7.4),采用1w的近紅外光激光發射器進行照射,每隔10min照射一次,取出2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。按照實施例1中相同的計算方式通過紫外分光光度法測定介質中藥物的濃度。
(3)按照實施例1相同的方式測試其抗菌性能,其抗菌效果與實施例1中結果相似。
對本實施例所得到的智能復合水凝膠載體進行近紅外光照射,其藥物緩釋情況如圖4所示。
經過分析計算,本實例聚多巴胺藥物負載量為14%,如圖(3)所示。在ph為5的緩沖液體系中,該凝膠材料達到最大緩釋率(70%)的時間約為36h;在ph為7.4的緩沖液體系中,該凝膠材料達到最大緩釋率(50%)的時間約為34h,均具有良好的藥物緩釋持續性。
實施例3
(1)將1mg/ml聚多巴胺10ml和5mg鹽酸四環素均勻混合,攪拌36h后,離心,將上層未負載上的藥物倒出,沉淀下來的成分用去離子水反復洗滌幾次,得到負載藥物的聚多巴胺分散液;
(2)將質量百分比濃度為2%的納米纖維素和上述負載一定藥物量的聚多巴胺分散液混合均勻,聚多巴胺的加入量為0.2wt%(相對于納米纖維素的質量),倒入成型器中,緩慢滴加質量百分比濃度為3%的氯酸鈣溶液(2ml),靜止24h,可以得到納米纖維素/聚多巴胺復合智能凝膠藥物緩釋材料。
檢測方法
(1)將所得到的藥物水凝膠制劑置于模擬人體生理環境的pbs緩沖液中(ph=5和ph=7.4),水平震蕩(50r/min),每隔1小時取2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。緩沖液要現配現用。按照實施例1中相同的計算方式通過紫外分光光度法測定介質中藥物的濃度。
(2)將制備的智能復合凝膠藥物緩釋材料置于模擬人體的生理環境pbs緩沖液中(ph=5和ph=7.4),采用4w的近紅外光激光發射器進行照射,每隔10min照射一次,取出2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。按照實施例1中相同的計算方式通過紫外分光光度法測定介質中藥物的濃度。
(5)按照實施例1相同的方式測試其抗菌性能,其抗菌性與實施例1中的抗菌效果相似。
對本實施例所得的智能復合水凝膠載體在不同的ph緩沖液體系中進行緩釋,藥物緩釋情況如圖5所示。
經過分析計算,本實例聚多巴胺藥物負載量為22%,如圖(3)所示。在ph為5的緩沖液體系中,該凝膠材料達到最大緩釋率(80%)的時間約為40h;在ph為7.4的緩沖液體系中,該凝膠材料達到最大緩釋率(65%)的時間約為39h,均具有藥物緩釋的持續性。
實施例4
(1)將1mg/ml聚多巴胺10ml和5mg鹽酸四環素均勻混合,攪拌48h后,離心,將上層未負載上的藥物倒出,沉淀下來的成分用去離子水反復洗滌幾次,得到負載藥物的聚多巴胺分散液;
(2)將質量百分比濃度為2%納米纖維素和上述負載一定藥物量的聚多巴胺分散液混合均勻,聚多巴胺的加入量為0.2wt%(相對于納米纖維素的質量),倒入成型器中,緩慢滴加質量百分比濃度為4%的氯化鈣溶液(2ml),靜止24h,可以得到納米纖維素/聚多巴胺復合智能凝膠藥物緩釋材料。
檢測方法
(1)將所得到的藥物水凝膠制劑置于模擬人體生理環境的pbs緩沖液中(ph=5和ph=7.4),水平震蕩(50r/min),每隔1小時取2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。緩沖液要現配現用。按照實施例1中相同的計算方式通過紫外分光光度計法測定介質中藥物的濃度。
(2)將制備的藥物緩釋材料置于模擬人體的生理環境pbs緩沖液中(ph=5和ph=7.4),采用5w的近紅外光激光發射器進行照射,每隔10min照射一次,取出2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。按照實施例1中相同的計算方式通過紫外分光光度法測定介質中藥物的濃度。
(3)按照實施例1相同的方式測試其抗菌性能,其抗菌效果與實施例1中結果相似。
經過分析計算,本實例聚多巴胺藥物負載量為10%,如圖(3)所示。在ph為5的緩沖液體系中,該凝膠材料達到最大緩釋率(82%)的時間約為36h;在ph為7.4的緩沖液體系中,該凝膠材料達到最大緩釋率(75%)的時間約為36h,均具有良好的藥物緩釋持續性。
實施例5
(1)將2mg/ml聚多巴胺10ml和6mg紫杉醇均勻混合,攪拌48h后,離心,將上層未負載上的藥物倒出,沉淀下來的成分用去離子水反復洗滌幾次,得到負載藥物的聚多巴胺分散液,其中聚多巴胺的sem和tem表征如圖7所示,由圖可以看出制備出的聚多巴胺的直徑為400nm左右,且分散均勻;
(2)將質量百分比濃度為5%納米纖維素和上述負載一定藥物量的聚多巴胺分散液混合均勻,聚多巴胺的加入量為0.3wt%(相對于納米纖維素的質量),倒入成型器中,緩慢滴加質量百分比濃度為6%的氯化鈣溶液(2ml),靜止20h,可以得到納米纖維素/聚多巴胺復合智能凝膠藥物緩釋材料。
檢測方法
(1)將所到的藥物水凝膠制劑置于模擬人體生理環境pbs緩沖液中(ph=5和ph=7.4),水平震蕩(50r/min),每隔1小時取2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。緩沖液要現配現用。按照實施例1中相同的計算方式通過紫外分光光度計法測定介質中藥物的濃度。
(2)將制備的智能復合凝膠藥物緩釋材料置于模擬人體的生理環境pbs緩沖液中(ph=5和ph=7.4),采用3w的近紅外光激光發射器進行照射,每隔10min照射一次,取出2ml溶液,并取相同量的緩沖液放回其中以維持總的溶液體積不變。按照實施例1中相同的計算方式通過紫外分光光度法測定介質中藥物的濃度。
經過分析計算,本實例聚多巴胺藥物負載量為15%。在ph為5的緩沖液體系中,該凝膠材料達到最大緩釋率(65%)的時間約為48h;在ph為7.4的緩沖液體系中,該凝膠材料達到最大緩釋率(45%)的時間約為48h,均具有良好的藥物緩釋持續性。
對比實施例1
(1)將1mg/ml聚多巴胺10ml和5mg鹽酸四環素均勻混合,攪拌12h后,離心,將上層未負載上的藥物倒出,沉淀下來的成分用去離子水反復洗滌幾次,得到負載藥物的聚多巴胺分散液;
(2)將質量百分比濃度為0.1%納米纖維素和上述負載一定藥物量的聚多巴胺分散液混合均勻,聚多巴胺的加入量為0.2wt%(相對于納米纖維素的質量),倒入成型器中,緩慢滴加質量百分比濃度為5%的氯化鈣溶液(2ml),靜止24h。
實驗結果表明:在納米纖維加入量為0.1%的情況下,得不到納米纖維素/聚多巴胺復合智能凝膠藥物緩釋材料,無法成型。
對比實施例2
(1)將1mg/ml聚多巴胺10ml和5mg鹽酸四環素均勻混合,攪拌2h后,離心,將上層未負載上的藥物倒出,沉淀下來的成分用去離子水反復洗滌幾次,得到負載藥物的聚多巴胺分散液;
(2)將質量百分比濃度為2%納米纖維素和上述負載一定藥物量的聚多巴胺分散液混合均勻,聚多巴胺的加入量為0.2wt%(相對于納米纖維素的質量),倒入成型器中,緩慢滴加質量百分比濃度為4%的葡萄糖酸鈣溶液(2ml),靜止24h,可以得到納米纖維素/聚多巴胺復合智能凝膠藥物緩釋材料。
檢測方法
(1)將所得到的藥物水凝膠制劑置于模擬人體的生理環境pbs緩沖液中(ph=5和ph=7.4),水平震蕩(50r/min),每隔1小時取2ml溶液,并取相同量的緩沖液放回其中以維持總的溶液體積不變。緩沖液要現配現用。按照實施例1中相同的計算方式通過紫外分光光度計法測定介質中藥物的濃度。
(2)將制備的智能復合凝膠藥物緩釋材料置于模擬人體生理環境pbs緩沖液中(ph=5和ph=7.4),采用1w的近紅外光激光發射器進行照射,每隔10min照射一次,取出2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。按照實施例1中相同的計算方式通過紫外分光光度法測定介質中藥物的濃度。
實驗結果表明:與實施例2相比,在聚多巴胺與藥物混合時間不充分的情況下,藥物負載量小于0.1%,導致凝膠材料緩釋效果不明顯。
對比實施例3
(1)將1mg/ml聚多巴胺10ml和5mg鹽酸四環素,攪拌48h后,離心,將上層未負載上的藥物倒出,沉淀下來的成分用去離子水反復洗滌幾次,得到負載藥物的聚多巴胺分散液;
(2)將質量百分比濃度為2%納米纖維素和上述負載一定藥物量的聚多巴胺分散液,聚多巴胺的加入量為10wt%(相對于納米纖維素的質量),混合均勻,倒入成型器中,緩慢滴加質量百分比濃度為4%的氯酸鈣溶液(2ml),靜止24h,得到納米纖維素/聚多巴胺復合凝膠藥物緩釋材料。
實驗結果表明:與實施例4相比,在聚多巴胺用量過多的情況下,納米纖維素/聚多巴胺復合凝膠藥物緩釋材料的強度很差(維持自身形狀的物理穩定性)不能應用于藥物緩釋。
對比實施例4
(1)將質量百分比濃度為2%納米纖維素2g和親水性藥物鹽酸四環素5mg均勻混合,攪拌36h后,倒入成型器中,緩慢滴加質量百分比濃度為3%的氯酸鈣溶液(2ml),靜止24h,可以得到納米纖維素凝膠藥物緩釋材料;
(2)將所得到的藥物水凝膠制劑置于模擬人體生理環境的pbs緩沖液中(ph=5和ph=7.4),水平震蕩(50r/min),每隔1小時取2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。按照實施例1中相同的計算方式計算藥物負載率和藥物緩釋率。
實驗結果表明,在ph為5的緩沖液體系中,該凝膠材料達到最大緩釋率(68%)的時間約為10h;在ph為7.4的緩沖液體系中,該凝膠材料達到最大緩釋率(50%)的時間約為9h,顯著低于實施例3(39-40h)。所以,純納米纖維水凝膠材料的藥物緩釋效果明顯不如本發明中制備的復合凝膠材料好。
對比實施例5
(1)將2mg/ml聚多巴胺10ml和6mg藥物紫杉醇均勻混合,攪拌48h后,離心,將上層未負載上的藥物倒出,沉淀下來的成分用去離子水反復洗滌幾次,得到負載藥物的聚多巴胺分散液;
(2)將上述負載藥物的聚多巴胺水分散液置于模擬人體生理環境的pbs緩沖液中(ph=5和ph=7.4),水平震蕩(50r/min),每隔1小時取2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。緩沖液要現配現用。通過紫外分光光度法測定介質中藥物的濃度。按照實施例1中相同的計算方式計算藥物緩釋率。
實驗結果表明:與實施例5相比,藥物緩釋過程出現明顯的突釋現象,不能達到藥物可控釋放的結果。證明聚多巴胺不能單獨用作藥物緩釋材料。
對比實施例6
(1)將1.5mg/ml聚多巴胺10ml和6mg鹽酸四環素均勻混合,攪拌36h后,離心,將上層未負載上的藥物倒出,沉淀下來的成分用去離子水反復洗滌幾次,得到負載藥物的聚多巴胺分散液。
(2)在上述制備得到的負載藥物的聚多巴胺分散液中加入0.266g異丙基丙烯酰胺和0.02gn,n’亞甲基雙(丙烯酰胺),聚多巴胺的加入量為0.2wt%(相對于異丙基丙烯酰胺和丙烯酰胺的總質量),在氮氣保護的情況下,低溫攪拌20分鐘。然后加入1.1mg過硫酸銨和1ul四甲基乙二胺。然后轉移到室溫環境下得到復合凝膠材料。
檢測方法
(1)將上述凝膠材料置于模擬人體生理環境的pbs緩沖液中(ph=5和ph=7.4),水平震蕩(50r/min),每隔1小時取2ml溶液,并取相同量的pbs緩沖液放回其中以維持總的溶液體積不變。緩沖液要現配現用。通過紫外分光光度法測定介質中藥物的濃度。按照實施例1中相同的計算方式計算藥物負載率和藥物緩釋率。
實驗結果表明:經過分析計算,本對比實施例的聚多巴胺藥物負載量為21%。在ph為5的緩沖液體系中,該凝膠材料達到最大緩釋率(50%)的時間約為20h;在ph為7.4的緩沖液體系中,該凝膠材料達到最大緩釋率(45%)的時間約為21h,顯著低于實施例3(39-40h)。因此,該凝膠材料的緩釋效果不如納米纖維素/聚多巴胺凝膠材料緩釋效果明顯,而且該凝膠材料制備過程復雜,制備環境苛刻,且生物相容性差。
根據本發明的納米纖維素和聚多巴胺均具有生物相容性,可完全降解,易于廢棄處理,有利于保護環境,是一種具有廣泛應用前景的納米纖維素藥物緩釋材料。而丙烯酰胺是石油基產品,不屬于綠色可持續性材料。
- 還沒有人留言評論。精彩留言會獲得點贊!