磁性復合顆粒及其制備方法和應用與流程
本發明屬于納米材料領域,涉及一種磁性復合顆粒及其制備方法和應用,具體涉及一種核殼衛星結構的磁性復合顆粒(AgMNP–AuNRs)及其制備方法和應用。
背景技術:
由于納米級粗糙的金屬表面對吸附在上面的探針分子具有巨大的拉曼增強效應,因此表面增強拉曼光譜(SERS)是一種非常重要的、可以應用到小分子痕量檢測的振動光譜分析技術。基于它的高靈敏以及指紋識別特性,SERS已經在很多領域得到了應用。SERS能夠得到應用,甚至未來可以在臨床診斷領域發揮作用的關鍵在于高性能SERS增強基底的可控制備。
在尺度為1~5nm的納間隙處會形成SERS“熱點”,處于SERS“熱點”區域的探針分子的拉曼信號會被強烈的放大。目前,雖然已經有許多帶有“熱點”結構的SERS增強基底被報道,但是大多數已經被報道的SERS“熱點”結構是零維,一維或者二維材料,SERS“熱點”的密度和數量都比較有限。
在過去的幾年間,將磁性內核和貴金屬納米顆粒復合在一起形成核殼衛星結構納米復合顆粒來構筑SERS基底的方法,引起廣泛的關注和研究。研究人員發現,這類基于磁性內核和貴金屬納米顆粒的復合物,相比傳統的膠體類SERS基底具有一些獨特的優勢。第一,它們非常適合檢測溶解在水中或者是乙醇中的化學物質,因為它們的超順磁性保證了它們可以在外加磁場的作用下,對目標化學物質做到很好的富集,而不用經過離心過程,可以避免離心損失,甚至是離心導致的沉降。第二,在外加磁場的作用下,這類磁性基底可以團聚在一起,就會形成顆粒間的“熱點”,從而進一步的增強SERS信號。在過去的十幾年間有許多研究人員研究了這類核殼衛星結構的磁性復合顆粒的構筑方法。比如,有的學者通過化學鍵(Au-S)直接連接Fe3O4和金納米顆粒來構筑Fe3O4–AuNPs磁性復合物。有的學者通過靜電方法,利用帶正電的Fe3O4來吸附帶負電的金納米顆粒來直接構筑這類Fe3O4–AuNPs磁性復合物。有的學者通過靜電方法,利用PEI修飾的Fe3O4(帶正電)來吸附PVP修飾的金納米棒(帶負電)構筑Fe3O4–AuNRs磁性復合物。但是,這類核殼衛星結構SERS增強基底的特點就是,磁性內核都沒有SERS增強能力,只是充當磁核,SERS增強由外圍連接的貴金屬納米顆粒保證,SERS增強效果有限。
技術實現要素:
本發明要解決的技術問題是克服現有技術的不足,提供一種具有很好的磁響應能力和SERS性能的磁性復合顆粒,還提供一種制備時間短、工藝簡單、通用性強、適用性廣的磁性復合顆粒的制備方法,并相應提供上述磁性復合顆粒作為SERS增強基底的檢測應用。
為解決上述技術問題,本發明采用以下技術方案:
一種磁性復合顆粒,所述磁性復合顆粒為核殼衛星結構,所述磁性復合顆粒包括銀殼磁珠和金納米棒,所述銀殼磁珠為磁性復合顆粒的磁性內核,所述銀殼磁珠表面通過聚(2-乙烯吡啶)與所述金納米棒結合;所述銀殼磁珠是由錳鐵氧體和連續包被在錳鐵氧體上的銀殼組成。
上述的磁性復合顆粒中,優選的,所述磁性復合顆粒的磁飽和值為10emu/g~35emu/g;和/或,所述銀殼磁珠的磁飽和值為40emu/g~50emu/g,所述錳鐵氧體的粒徑為100nm~300nm,所述銀殼的厚度為15nm~30nm。
作為一個總的發明構思,本申請還提供一種磁性復合顆粒的制備方法,包括以下步驟:
(1)將銀殼磁珠加入到聚(2-乙烯吡啶)溶液中,經超聲處理后,在銀殼磁珠表面形成聚(2-乙烯吡啶)多聚物層,用磁鐵富集,得到聚(2-乙烯吡啶)修飾的銀殼磁珠;
(2)制備金納米棒溶液;
(3)將步驟(1)所得聚(2-乙烯吡啶)修飾的銀殼磁珠與步驟(2)制備的金納米棒溶液進行混合,保證金納米棒過量,經超聲反應后,通過磁富集,得到磁性復合顆粒。
上述的磁性復合顆粒的制備方法中,優選的,所述步驟(1)中,所述聚(2-乙烯吡啶)溶液中聚(2-乙烯吡啶)的濃度為2mg/mL~10mg/mL,所述聚(2-乙烯吡啶)溶液的溶劑為乙醇;和/或,所述銀殼磁珠與所述聚(2-乙烯吡啶)溶液的比值為5mg~20mg∶2mL~6mL;和/或,所述超聲處理的時間為30min~60min。
上述的磁性復合顆粒的制備方法中,優選的,所述步驟(1)中,所述銀殼磁珠的制備方法包括以下步驟:
(1.1)將錳鐵氧體加入到聚醚酰亞胺溶液中,超聲,在錳鐵氧體表面形成帶正電的聚醚酰亞胺多聚物層,用磁鐵富集,得到聚醚酰亞胺修飾的錳鐵氧體;
(1.2)將聚醚酰亞胺修飾的錳鐵氧體加入到帶負電的金種子溶液中,超聲,使所述聚醚酰亞胺修飾的錳鐵氧體的表面吸附上帶負電的金種子,然后用磁鐵富集,得到中間產物;
(1.3)將步驟(1.2)所得的中間產物超聲分散于聚乙烯吡咯烷酮溶液中,加入硝酸銀溶液,超聲混合,再加入甲醛溶液和氨水溶液進行超聲反應,最后用磁鐵富集,得到銀殼磁珠。
上述的磁性復合顆粒的制備方法中,優選的,所述步驟(1.1)中,所述聚醚酰亞胺溶液中聚醚酰亞胺的濃度為2mg/mL~5mg/mL;所述錳鐵氧體與所述聚醚酰亞胺溶液的比值為0.1g∶50mL;所述超聲的時間為10min~30min;
和/或,所述步驟(1.2)中,所述金種子溶液主要由氯金酸溶液、檸檬酸鈉溶液和硼氫化鈉溶液加入水中攪拌制備得到;
和/或,所述步驟(1.3)中,所述聚乙烯吡咯烷酮溶液中聚乙烯吡咯烷酮的濃度為1.0mg/mL~1.5mg/mL;所述硝酸銀溶液中硝酸銀的濃度為2mg/mL~4mg/mL;所述甲醛溶液中甲醛的濃度為36wt%~38wt%;所述氨水溶液中氨水的濃度為25wt%~28wt%;所述中間產物、聚乙烯吡咯烷酮溶液、硝酸銀溶液、甲醛溶液和氨水溶液的比值為10mg∶200mL∶1mL∶150μL∶100μL~500μL。
上述的磁性復合顆粒的制備方法中,優選的,所述步驟(2)中,所述金納米棒溶液的制備方法包括以下步驟:
(2.1)將氯金酸溶液加入到十六烷基三甲基溴化銨溶液中,在恒溫攪拌條件下,加入硼氫化鈉溶液,經反應后,停止攪拌,繼續恒溫老化,得到金種子溶液;
(2.2)將氯金酸溶液加入到十六烷基三甲基溴化銨溶液中,然后加入硝酸銀溶液和鹽酸溶液,再加入抗壞血酸溶液,最后加入金種子溶液,在恒溫水浴下進行生長,得到初步的金納米棒溶液;
(2.3)將初步的金納米棒溶液進行離心處理,然后用水重懸,得到金納米棒溶液。
上述的磁性復合顆粒的制備方法中,優選的,所述步驟(2.1)中,所述氯金酸溶液的濃度為0.5mM,所述十六烷基三甲基溴化銨溶液的濃度為0.2M,所述硼氫化鈉溶液的濃度為0.01M,所述氯金酸溶液、十六烷基三甲基溴化銨溶液、硼氫化鈉溶液的體積比為2.5mL∶2.5mL∶0.3mL;所述恒溫攪拌的溫度為29℃,所述反應的時間為1min,所述恒溫老化的溫度為29℃,所述恒溫老化的時間為1h~2h;
和/或,所述步驟(2.2)中,所述氯金酸溶液的濃度為1mM,所述十六烷基三甲基溴化銨溶液的濃度為0.2M,所述硝酸銀溶液的濃度為0.01M,所述鹽酸溶液的濃度為2M,所述抗壞血酸溶液的濃度為0.1M;所述氯金酸溶液、十六烷基三甲基溴化銨溶液、硝酸銀溶液、鹽酸溶液、抗壞血酸溶液、金種子溶液的體積比為20mL∶20mL∶0.4mL∶0.38mL∶0.32mL∶140μL;所述恒溫水浴的溫度為29℃,所述生長的時間為10h~16h;
和/或,所述步驟(2.3)中,所述初步的金納米棒溶液的離心次數為3次~5次,離心轉速為7500rpm~8500rpm,離心時間為6min~7min。
上述的磁性復合顆粒的制備方法中,優選的,所述步驟(3)中,所述超聲反應的時間為30min~60min。
作為一個總的發明構思,本申請還提供一種上述的磁性復合顆粒或上述的制備方法制得的磁性復合顆粒在作為表面增強拉曼散射增強基底中的應用。
本發明的步驟(1.1)中,錳鐵氧體的制備過程如下:
將含鐵的前驅體和含錳的前驅體溶解于溶劑中,加入表面活性劑,然后油浴加熱至表面活性劑溶解,停止加熱,加入靜電穩定劑并攪拌,再進行溶劑熱合成反應,冷卻后,用磁鐵富集反應產物,得到錳鐵氧體;
所述含鐵的前驅體為FeCl3·6H2O,所述含錳的前驅體為MnCl2·4H2O,所述溶劑為乙二醇,所述表面活性劑為聚乙烯吡咯烷酮,所述靜電穩定劑為醋酸鈉;所述FeCl3·6H2O、MnCl2·4H2O、乙二醇、聚乙烯吡咯烷酮和醋酸鈉的比值為360mg∶131.94mg∶20mL∶2g∶1.5g;
更優選的,所述油浴加熱的溫度為110℃~130℃,加熱時間為10min~20min;所述溶劑熱合成反應的溫度為200℃~210℃,反應時間為10h~16h。
本發明的步驟(1.2)中,金種子溶液制備時,更優選的,所述氯金酸溶液中氯金酸濃度為1wt%;所述檸檬酸鈉溶液中檸檬酸鈉濃度為1wt%;所述硼氫化鈉溶液中硼氫化鈉的濃度為0.1M;所述氯金酸溶液、檸檬酸鈉溶液、硼氫化鈉溶液和水的體積比為3.4∶2.94∶12∶400。
本發明的磁性復合顆粒的應用中,更優選的,所述應用包括磁性復合顆粒在檢測生物化學物質中的應用,所述生物化學物質能與金或銀表面通過化學鍵結合,包括以下步驟:
將所述磁性復合顆粒加入到含生物化學物質的甲醇溶液中超聲反應,用磁鐵富集,得到反應后的磁性復合顆粒;將反應后的磁性復合顆粒重懸于乙醇中,點至硅片上進行光譜測試,得到拉曼測試信號,利用所述拉曼測試信號對所述生物化學物質的濃度進行定性或定量檢測。
本發明中,磁性復合顆粒可寫作AgMNP-AuNRs,銀殼磁珠為AgMNPs,金納米棒為AuNRs。
本發明中,濃度M即為mol/L,mM即為mmol/L。
本發明為了進一步提高SERS增強效果,提出了一種新型的三維(3D)核殼衛星結構的磁性復合顆粒,以銀殼磁珠為磁性內核,以金納米棒為衛星納米顆粒構筑了三維核殼衛星結構磁性復合顆粒。這種結構的內核和衛星之間形成納間隙,構成高密度的SERS“熱點”,與磁引誘導致的團聚形成的顆粒間的“熱點”一起進一步增強目標分子的拉曼信號,顯著增強該區域目標分子的拉曼信號。本發明磁性內核和衛星顆粒都有SERS增強能力,在銀殼磁珠和衛星金納米棒之間形成了高密度的“熱點”結構。本發明制備的磁性復合顆粒非常適用于生化小分子的高靈敏SERS檢測。
與現有技術相比,本發明的優點在于:
(1)本發明的磁性復合顆粒具有核殼衛星結構,磁性內核為銀殼磁珠(AgMNPs),衛星納米顆粒為金納米棒(AuNRs),具有很好的磁響應能力以及SERS性能。磁響應能力由超順磁性的內核保證,SERS性能由銀殼磁珠和金納米棒共同保證。
(2)本發明的磁性復合顆粒通過共價鍵法構筑,利用P2VP層連接銀殼磁珠和金納米棒,這種結構的內核和衛星之間形成納間隙,納間隙尺寸在1nm以下,構成高密度的SERS“熱點”,可以顯著增強該區域目標分子的拉曼信號。
(3)本發明的磁性復合顆粒的制備方法的關鍵在于P2VP分子上有許多吡啶基團,這些吡啶基團可以通過氮原子提供孤對電子與金屬表面形成共價鍵。因此,只需要P2VP這一種功能分子就可以實現核殼衛星結構的AgMNP-AuNRs的構筑。當P2VP修飾到銀殼磁珠表面之后,還有許多空余的吡啶基團可以繼續連接金納米棒。
(4)本發明的磁性復合顆粒的制備方法,磁性內核是具有超順磁性、具有良好SERS性能的銀殼磁珠,并且銀殼磁珠的制備是以錳鐵氧體為內核通過超聲輔助種子生長法實現的。且可以根據需要,通過控制硝酸銀溶液的加入量,在不同粒徑的錳鐵氧體上包被任意厚度的銀殼。因而本發明的制備方法通用性強、適應性廣。
(5)本發明的磁性復合顆粒的應用,該磁性復合顆粒用作生化傳感器對生物化學物質進行檢測時,尤其是對含有巰基或者二硫鍵這類可以與金或銀表面形成化學鍵的物質時,可以通過磁富集對樣品進行濃縮,檢測靈敏度高,操作方便,而且有望為人體激素、病原體、殘留化學毒素等生物化學物質的檢測提供便利,尤其是在低濃度生物化學物質SERS檢測上,提供快速、實用、高靈敏的SERS增強基底。
綜上,本申請通過共價鍵連接(P2VP)的方法,實現了核殼衛星結構的磁性復合顆粒的可控制備,反應簡單,操作方便,制備的磁性復合顆粒,具有很好的SERS活性以及磁響應能力。在生物樣品分離以及生物醫學檢測應用等領域都有很好的應用前景。
附圖說明
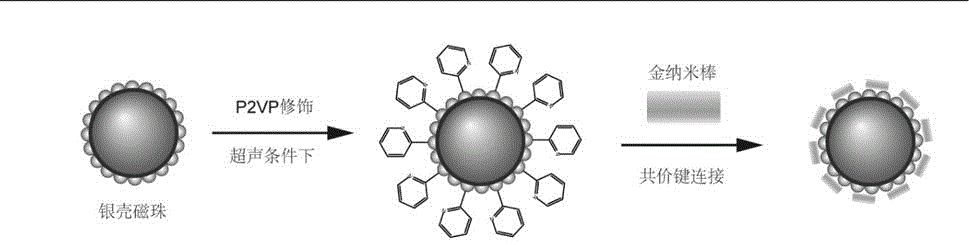
圖1為本發明的磁性復合顆粒(AgMNP-AuNRs)的制備方法示意圖。
圖2為本發明實施例1制得的MnFe2O4、MnFe2O4@PEI-auseeds和銀殼磁珠的透射電子圖,其中,(a)圖為MnFe2O4;(b)圖為MnFe2O4@PEI-auseeds;(c)圖為銀殼磁珠。
圖3為本發明實施例1制得的MnFe2O4、MnFe2O4@PEI-auseeds和銀殼磁珠的XRD圖譜。
圖4為本發明實施例1制得的MnFe2O4、MnFe2O4@PEI-auseeds和銀殼磁珠的磁響應曲線。
圖5為本發明實施例1制得的銀殼磁珠的掃描電鏡圖。
圖6為本發明實施例1制得的金納米棒的透射電子顯微鏡照片。
圖7為本發明實施例1制得的金納米棒的紫外-可見吸收光譜圖。
圖8為本發明實施例1制得的磁性復合顆粒(AgMNP-AuNRs)的掃描電子顯微鏡照片(a和b),透射電子顯微鏡照片(c)以及高分辨率透射電子顯微鏡照片(d)。
圖9為本發明實施例1制得的磁性復合顆粒(AgMNP-AuNRs)的磁滯曲線圖。
圖10為本發明實施例1制得的磁性復合顆粒(AgMNP-AuNRs)用于檢測Thiram的檢測流程圖。
圖11為本發明實施例1制得的磁性復合顆粒(AgMNP-AuNRs)檢測Thiram的光譜圖。
圖12為本發明實施例1制得的磁性復合顆粒(AgMNP-AuNRs)檢測Thiram的強度-濃度校正曲線圖。
具體實施方式
以下結合說明書附圖和具體優選的實施例對本發明作進一步描述,但并不因此而限制本發明的保護范圍。
以下實施例中所采用的材料和儀器均為市售。
實施例1:
一種本發明的磁性復合顆粒(具體為AgMNP-AuNRs),該磁性復合顆粒為核殼衛星結構,包括銀殼磁珠和金納米棒,銀殼磁珠為磁性復合顆粒的磁性內核,銀殼磁珠表面通過聚(2-乙烯吡啶)與金納米棒結合,銀殼磁珠是由錳鐵氧體和連續包被在錳鐵氧體上的銀殼組成。
本實施例中,所述磁性復合顆粒的磁飽和值為31.1emu/g,磁性內核為超順磁性的銀殼磁珠,粒徑為360nm,其中錳鐵氧體的粒徑為300nm,銀殼的厚度為30nm,銀殼磁珠的磁飽和值為46.4emu/g。銀殼磁珠外圍的金納米棒尺寸為72×18nm,LSPR共振峰為840nm。
一種上述本實施例的磁性復合顆粒(AgMNP-AuNRs)的制備方法,如圖1所示,包括以下步驟:
(1)制備銀殼磁珠
(1.1)制備單分散超順磁性的錳鐵氧體(MnFe2O4)
將(4/3mmol, 360 mg)FeCl3·6H2O和(2/3mmol, 131.94 mg)MnCl2·4H2O通過磁力攪拌溶解到20mL乙二醇(EG)中,加入2g聚乙烯吡咯烷酮(PVP),轉移到120℃的油浴中,加熱10min,直到溶液呈透明狀,停止加熱;再加入1.5g醋酸鈉,轉移到磁力攪拌器上,攪拌30min,得到比較均勻的混合溶液后轉移到50mL的聚四氟乙烯反應釜中。置入提前加熱到200℃的烘箱中,反應10h后取出,冷卻至室溫,用磁鐵富集反應產物,將反應產物用乙醇清洗3~5次,再用水清洗3~5次。在60℃的真空烘箱中烘干,得到粒徑為300nm的錳鐵氧體(MnFe2O4),備用。
本步驟中,利用乙二醇作為溶劑的溶劑熱法制備超順磁性的錳鐵氧體,作為銀殼磁珠的磁性內核。制備過程中,三氯化鐵和氯化錳被用作前驅體,而EG既充當溶劑又充當還原劑。聚乙烯吡咯烷酮作為表面活性劑,醋酸鈉被用作靜電穩定劑,防止顆粒團聚。
(1.2)制備PEI修飾的錳鐵氧體(MnFe2O4@PEI)
稱取0.1g錳鐵氧體加入到50mL聚醚酰亞胺(PEI)水溶液中,其中,聚醚酰亞胺的濃度為5mg/mL,超聲20min,利用靜電結合作用,在MnFe2O4表面形成一層厚度為2nm的帶正電的PEI多聚物層,后用磁鐵富集清洗,得到聚醚酰亞胺修飾的錳鐵氧體(MnFe2O4@PEI)。
通過超聲時間可以控制PEI多聚物層的厚度,PEI多聚物層的厚度優選為1nm~3nm,不宜過厚,否則會影響產物的磁響應特性,也會影響產物的表面形貌。
(1.3)制備金種子粒徑為4nm的帶負電的金種子溶液
在500mL藍蓋瓶中加入400mL水,并開始以950rpm的轉速進行磁力攪拌;先后加入3.4mL濃度為1wt%的HAuCl4溶液和2.94mL濃度為1wt%的檸檬酸鈉溶液;再加入新制的12mL濃度為0.1M的硼氫化鈉溶液,室溫攪拌4h后,得到金種子粒徑為4nm的帶負電的金種子溶液,備用。
(1.4)制備吸附金種子的聚醚酰亞胺修飾的錳鐵氧體(MnFe2O4@PEI-auseeds)
將0.1g MnFe2O4@PEI 加入到200mL步驟(1.3)所得的帶負電的金種子溶液中超聲1h,用磁鐵富集清洗后,得到中間產物MnFe2O4@PEI-auseeds。該MnFe2O4@PEI-auseeds可以長期存放(1~3個月)。
4 nm金種子的量優選為200mL~300mL,只要保證金種子過量即可。
(1.5)制備銀殼磁珠
將步驟(1.4)所得的中間產物超聲分散于聚乙烯吡咯烷酮溶液中,加入硝酸銀溶液,超聲混合,再加入甲醛溶液和氨水溶液進行超聲反應,最后用磁鐵富集,得到銀殼磁珠。具體過程如下:
利用超聲輔助種子生長法在MnFe2O4@PEI-auseeds上包被銀殼,將10mg中間產物MnFe2O4@PEI-auseeds加入到200mL濃度為1.5mg/mL的聚乙烯吡咯烷酮水溶液中,超聲2min;加入1mL硝酸銀溶液(2mg/mL),超聲2min;加入150μL濃度為36-38wt%的甲醛溶液,再加入300μL濃度為25-28wt%的氨水溶液超聲1min后,用磁鐵富集得到銀殼磁珠。
(2)銀殼磁珠P2VP修飾
在超聲條件下,將20mg聚(2-乙烯吡啶)(P2VP)溶解到4mL乙醇中,形成5mg/mL的P2VP溶液,然后加入10mg銀殼磁珠,超聲處理1h,再利用磁富集的手段,用乙醇清洗5次,去除過量的P2VP,即得到P2VP修飾的銀殼磁珠。
(3)金納米棒的制備
(3.1)將2.5mL、濃度為0.5mM的氯金酸溶液加入到2.5mL、0.2M的十六烷基三甲基溴化銨(CTAB)溶液中,在500rpm、29℃的恒溫磁力攪拌條件下,向上述溶液中加入300μL、0.01M新配置的NaBH4溶液,1min后,溶液呈棕黃色,關閉磁力攪拌,將得到的種子液在29℃下繼續老化1h,得到金種子溶液,備用;
(3.2)將20mL濃度為1mM的氯金酸溶液加入到20mL、0.2M十六烷基三甲基溴化銨溶液中,輕微搖勻后,向溶液中加入0.4mL、0.01M的硝酸銀溶液,輕微搖勻后,加入0.38mL、2M鹽酸溶液,再加入0.32mL、0.1M抗壞血酸(AA)溶液后,輕微搖勻,溶液呈無色,最后加入140μL上述步驟(3.1)得到的金種子溶液后輕微搖勻,置于預先設置到29℃的恒溫水浴中,生長12h,得到初步的金納米棒溶液;
(3.3)將初步的金納米棒溶液離心3次,離心速度為7500rpm,每次的離心時間為6min,盡可能的去除金納米棒表面的十六烷基三甲基溴化銨,然后用水重懸(可以重懸到10 mL去離子水中,得到4倍濃縮的金納米棒溶液,即4×AuNRs),備用。
(4)核殼衛星結構的磁性復合顆粒(AgMNP-AuNRs)的制備
將步驟(2)所得P2VP修飾過的銀殼磁珠與步驟(3)所得的經過三次離心處理的金納米棒進行混合,保證金納米棒過量,超聲反應30min,兩者通過共價鍵結合,形成核殼衛星結構的AgMNP-AuNRs磁性復合顆粒。然后,通過磁富集的手段,用去離子水清洗三次,去除過量的金納米棒,得到核殼衛星結構的AgMNP-AuNRs磁性復合顆粒。
圖2為本實施例步驟(1.1)制得的MnFe2O4、步驟(1.4)制得的MnFe2O4@PEI-auseeds和步驟(1.5)制得的銀殼磁珠的透射電子圖。其中,(a)圖為MnFe2O4的透射電子圖,由圖可見,所得的錳鐵氧體粒徑均一,單分散性好;(b)圖為MnFe2O4@PEI-auseeds的透射電子圖,(c)圖為銀殼磁珠的透射電子圖,由圖可見,納米級粗糙的銀殼連續地包被在磁性內核上,此結構能賦予銀殼磁珠優異的SERS性能。圖3為本實施例步驟(1.1)制得的MnFe2O4、步驟(1.4)制得的MnFe2O4@PEI-auseeds和步驟(1.5)制得的銀殼磁珠的XRD圖譜,其中,(a)為MnFe2O4;(b)為MnFe2O4@PEI-auseeds;(c)為銀殼磁珠。從圖3中的曲線a可以清楚地觀察到合成的MnFe2O4的衍射峰(圖中的實心方框)在29.6,35.1,42.5,52.6,56.1和61.6度,分別對應于MnFe2O4的(220),(311),(400),(422),(511)和(440)晶面。金種子被吸附到MnFe2O4上之后,在38.2度處(圖中的實心圓形)出現了一個新的對應于金的(111)晶面的X射線衍射峰(圖3的曲線b)。當連續的銀殼形成之后,我們在2θ值38.1,44.3,64.4,77.5,和81.5處又觀察到了五個新的衍射峰(圖3的曲線c),分別對應于銀的(111),(200),(220),(311)和(222)晶面(實心倒三角)。
圖4為本實施例步驟(1.1)制得的MnFe2O4、步驟(1.4)制得的MnFe2O4@PEI-auseeds和步驟(1.5)制得的銀殼磁珠的磁響應曲線,其中,(a)為MnFe2O4;(b)為MnFe2O4@PEI-auseeds;(c)為銀殼磁珠。首先,圖4中沒有出現磁滯回線,也就說明了制備得到的所有磁性納米顆粒都是超順磁性的。而這種超順磁性的特性可以防止納米顆粒的團聚,也能夠使得這些磁性納米顆粒在沒有外加磁場作用時,很好的分散以及重懸到溶液中。這種特性也方便了它們在生物樣品分離以及檢測方面的應用。如圖4所示,制備得到的銀殼磁珠的磁飽和值大約是46.4emu/g(曲線c)。
本實施例所制得的銀殼磁珠的掃描電鏡照片如圖5所示,由圖可見,所制備的銀殼磁珠外殼比較粗糙并且連續,是由彼此相鄰的較大的銀顆粒構成的。
圖6和圖7分別為本實施例步驟(3)制得的金納米棒的透射電子顯微鏡照片和紫外-可見吸收光譜。從圖6可以看出,制備得到的金納米棒尺寸比較均一,大顆粒較少,長徑比大約為4∶1。從圖7可以看出,制備得到的金納米棒的SPR共振峰大約為840nm。
圖8為本實施例步驟(4)制得的核殼衛星結構的磁性復合顆粒(AgMNP-AuNRs)的掃描電子顯微鏡照片(a和b),透射電子顯微鏡照片(c)以及高分辨率透射電子顯微鏡照片(d)。從圖中可以看出金納米棒是被成功連接在銀殼磁珠表面,在兩者連接處幾乎觀察不到CTAB和P2VP分子層,說明兩者之間可以形成間隙為1nm左右的納間隙。
圖9為本實施例步驟(4)制得的磁性復合顆粒(AgMNP-AuNRs)的磁滯曲線。從圖中可以看出,在連接金納米棒之后,磁飽和值由46.4emu/g降到了31.1emu/g。磁飽和值的逐步降低也說明了非磁性材料的成功包覆,這也是和電鏡表征的結果是相互印證的。
一種上述本實施例的磁性復合顆粒(AgMNP-AuNRs)作為SERS增強基底在對農殘福美雙(Thiram)檢測中的應用,如圖10所示,包括以下步驟:
(1)將本實施例制得的AgMNP-AuNRs分散在乙醇中,得到濃度為10mg/mL的AgMNP-AuNRs分散液;將6個不同濃度(10-6 M、10-7 M、10-8 M、10-9 M、10-10 M和10-11 M)的1mL福美雙的甲醇溶液分別加入到6個1.5mL的離心管中,然后每個離心管中加入5μL上述的磁性復合顆粒分散液,置入超聲中反應1h。
(2)用磁鐵富集反應后的AgMNP-AuNRs,并用乙醇清洗3~5次后,重懸到5μL乙醇中,用移液槍將重懸的反應后的AgMNP-AuNRs點到硅片上,晾干后進行光譜測試,利用得到的拉曼測試信號即可對待測樣品中的福美雙濃度進行定性或定量檢測。
所得的測試結果如圖11和圖12所示,圖11為拉曼光譜圖,圖12為強度-濃度校正曲線圖。由圖11可見,福美雙的主峰(1383 cm-1)在10-10M濃度以上都清晰可見。由此可見,制備得到的磁性復合顆粒對福美雙的檢測限是10-10M。圖12說明福美雙的濃度和SERS強度之間存在比較好的校正曲線關系,也就是說該磁性復合顆粒檢測福美雙的動態范圍為10-6M~10-10M。
以上所述僅是本發明的優選實施方式,本發明的保護范圍并不僅局限于上述實施例。凡屬于本發明思路下的技術方案均屬于本發明的保護范圍。應該指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下的改進和潤飾,這些改進和潤飾也應視為本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!