嵌合抗原受體及制備方法與流程
本申請要求2014年2月14日提交的美國臨時專利申請No.61/940,339的權益,所述申請通過引用方式將其全文并入本文。
發明領域
本發明一般涉及分子生物學和醫學領域。更具體地,它涉及產生嵌合抗原受體(CAR)的方法。
相關領域的描述
過繼性T細胞轉移是一種可以用于治療癌癥的有希望的治療方法。過繼性T細胞轉移牽涉分離并擴增能選擇性殺傷腫瘤細胞的抗原特異性T細胞。一般地,從受試者取出T細胞,并且在體外培養。可以將嵌合抗原受體(CAR)在體外引入T細胞中以指導T細胞在再引入受試者中后基于抗原的表達而選擇性殺傷腫瘤細胞(例如Wieczorek等2013;Berry等,2013)。
一個與過繼性T細胞轉移有關的問題是哪種CAR可以在某些患者群體中更有效起作用(例如用于治療特定癌癥)之間存在顯著的可變性。由于可以潛在產生的可以展現出針對癌癥的治療活性的潛在不同CAR的數目非常大,目前臨床醫生非常難以預期哪種CAR可以展示針對給定癌癥或癌癥亞型的治療活性。由于過繼性T細胞轉移的重大的治療潛力,明顯需要用于鑒定和產生新CAR的改善的方法。
技術實現要素:
在一些方面,本發明提供了用于產生CAR的方法,并且提供了特定的CAR。在一些方面,提供了用于產生大量CAR的方法,所述CAR可以針對特定癌癥或癌癥亞型的活性被篩選;這樣,可以產生并鑒定可以展現出改善的針對特定癌癥或癌癥亞型的治療潛力的CAR。可以向受試者或人患者治療性施用本文中提供的CAR,例如以治療癌癥。
臨床數據證明了使T細胞靶向給定的腫瘤相關抗原(TAA)的特定的嵌合抗原受體(CAR)設計在不同患者中可以具有不同的治療潛力。例如,當向具有急性的而不是慢性的B譜系白血病的患者施用自體遺傳修飾的T細胞時,經由嵌合CD28/CD3ζ或CD137/CD3-ζ活化的第二代CD19特異性CAR可以展現出優越的臨床響應。為了解決此問題,本文中提供了用于產生可以展現出改善的針對給定腫瘤的抗腫瘤效果的CAR種類的方法。
例如,本文中提供了可以用于產生大量CAR并且篩選它們治療來自給定患者的癌癥的能力的方法;這樣,可以使用所述方法使用于患者的療法個體化,并且選擇展示針對具有特定癌癥的特定患者或患者亞組的改善的治療潛力的特定CAR。用于基因療法的臨床方法可以利用來自睡美人(Sleeping Beauty)(SB)轉座子系統的DNA質粒的電轉移,例如以降低制造用于小患者亞組的單獨CAR設計的成本和復雜性。用于使CAR+T細胞個體化的這些方法可以利用大量CAR分子的產生,可以篩選所述CAR分子和評估其有益于給定患者的能力。
在一些方面,提供了使用三重位點特異性重組系統(又稱為“EZ-CAR”平臺)高通量裝配CAR分子的方法。在一些實施方案中,這些方法可以允許從(i)限定特異性的單鏈可變片段(scFv),(ii)從細胞表面附加scFv的支架/鉸鏈,和(iii)一個或多個胞內信號傳導結構域快速組合原型CAR的3種組分。例如,如下文實施例中顯示,使用EZ CAR平臺連同臨床級CD19RCD28mζCAR+T細胞(CG CAR),產生經由嵌合CD28/CD3-ζ活化的CD19特異性CAR。
在一些實施方案中,本文中提供或通過根據本發明的方法產生的CAR可以在T細胞中與膜結合IL-15共表達。這樣,T細胞可以在體外或體內在不顯著增殖的情況下以靜止狀態存活或存在。比較而言,如先前描述,表達CAR的T細胞當在體外去除細胞因子時通常會死亡,并且此細胞死亡可以在某些情況中臨床施用T細胞時充當安全性特征。通常使用自發細胞測定來測量T細胞增殖。因此,與某些先前鑒定的CAR(其中T細胞在沒有抗原性刺激的情況下在體外不能存活)形成對比,本文中提供了可以在體外在沒有自發生長的情況下誘導細胞毒性的CAR。根據期望的具體的實施方案,通過本發明的方法產生或本文中提供的CAR可以在T細胞中在具有或沒有膜結合IL-15在T細胞中共表達的情況下表達。
本發明的一個方面涉及一種組合物,所述組合物包含(a)編碼一個或多個獨特的抗原結合結構域的多個第一載體;(b)編碼一個或多個獨特的鉸鏈結構域的多個第二載體;和(c)編碼一個或多個獨特的膜內結構域的多個第三載體;其中所述第一、第二和第三載體中的至少兩種包括多個分別編碼獨特的抗原結合結構域、鉸鏈結構域和/或膜內結構域的兩種或更多種載體,并且進一步其中所述載體包含用于同源重組的位點以允許編碼嵌合抗原受體(CAR)的第四載體的產生。
在本發明中,如提到蛋白質結構域和多肽,如抗原結合結構域、鉸鏈結構域、跨膜結構域、和膜內結構域(endodomain)使用,術語“獨特的”意味著具有不同的多肽(氨基酸)序列、包含不同的多肽(氨基酸)序列或由不同的多肽(氨基酸)序列組成的結構域。例如,兩個“獨特的”抗原結合結構域可以結合相同抗原(實際上,甚至所述抗原上的相同表位);然而,抗原結合結構域在它們的連續氨基酸組成彼此不同的情況下是“獨特的”。同樣地,在連續的氨基酸組成方面不同的兩種“獨特的”抗原結合結構域也可以特異性結合不同抗原和表位。相反,如本文中使用,相同氨基酸序列的兩個分子(多肽)不是“獨特的”多肽。
在一些實施方案中,多個第一載體編碼多個獨特的抗原結合結構域,多個第二載體編碼一個鉸鏈結構域,并且多個第三載體編碼多個獨特的膜內結構域。在一些實施方案中,多個第一載體編碼多個獨特的抗原結合結構域,多個第二載體編碼多個獨特的鉸鏈結構域,并且多個第三載體編碼多個獨特的膜內結構域。在一些實施方案中,多個第一載體編碼多個獨特的抗原結合結構域,多個第二載體編碼多個獨特的鉸鏈結構域,并且多個第三載體編碼一個膜內結構域。在一些實施方案中,多個第一載體編碼一個抗原結合結構域,多個第二載體編碼多個獨特的鉸鏈結構域,并且多個第三載體編碼多個獨特的膜內結構域。在一些實施方案中,抗原結合結構域包含scFv或由scFv組成。第三載體可以編碼跨膜結構域。第二載體可以編碼跨膜結構域。在一些實施方案中,組合物進一步包含編碼一個或多個跨膜結構域的多個第五載體;其中第一載體、第二載體、第三載體和第五載體包含用于同源重組的位點以產生編碼嵌合抗原受體(CAR)的第四載體。第一載體可以包含同源重組的第一序列和第二位點。第二載體可以包含同源重組的第二序列和同源重組的第三序列。第三載體可以包含同源重組的第三序列和同源重組的第四序列。第三載體可以包含同源重組的所述第三序列和同源重組的第四序列。第四載體包含同源重組的第一序列和同源重組的第四序列。第一載體、第二載體、和/或第三載體可以編碼轉座酶。轉座酶可以是鮭魚型Tc1樣轉座酶(SB)。在一些實施方案中,第一載體、第二載體、第三載體、第四載體和/或第五載體中的1種、2種、3種、4種或全部是睡美人(SB)或piggyBac轉座子載體。或者,在一些實施方案中,第一載體、第二載體、第三載體、第四載體、和/或第五載體不是睡美人(SB)或piggyBac轉座子載體;例如,在一些實施方案中,可以在不使用睡美人(SB)或piggyBac載體的情況下產生CAR,然后可以隨后將CAR插入在適合于轉染T細胞的載體中(例如,插入睡美人(SB)載體,如例如Singh等,2015中所述)。不過,在一些實施方案中,產生已經存在于適合于轉染T細胞的載體中的CAR可以簡化過程或者減少產生CAR和轉染T細胞兩者所需的步驟的數目。獨特的抗原結合結構域可以選擇性結合不同的抗原。在一些實施方案中,獨特的抗原結合結構域選擇性結合相同的抗原。抗原結合結構域可以選擇性結合CD19、通用抗原(小鼠)、HER-3、GD2、Gp75、CS1蛋白、間皮素、磷脂酰絲氨酸、cMyc、CD22、CD4、CD44v6、CD45、CD28、CD3、CD3e、CD123、CD138、CD52、CD56、CD74、CD30、Gp75、CD38、CD33、CD20、Her1/HER3融合物、GD2、碳水化合物、曲霉、ROR1、c-MET、EGFR、Dectin、埃博拉、真菌、GP、HERV-K(HERVK)、NY-ESO-1、VEGF-R2、TGF-b2R、IgG4、生物素或O-AcGD2。獨特的抗原結合結構域可以由scFv組成或者包含scFv。鉸鏈區可以由下列各項組成或包含下列各項:12AA肽(GAGAGCAAGTACGGCCCTCCCTGCCCCCCTTGCCCT,SEQ ID NO:1)、t-20AA肽、IgG4FcΔ EQ、IgG4FcΔQ、(t-12AA+t-20AA)、mKate、phiLov、dsRed、Venus、eGFP、CH3HA,(CD8α+t-20AA)、雙重t-20AA、(t-20AA+CD8α)、(CD8α+亮氨酸拉鏈Basep1)、(CD8α+亮氨酸拉鏈Acid1)、2D3、CD8α或IgG4Fc。膜內結構域中的至少一個可以包含CD3ζ。膜內結構域中的至少一個可以包含一個或多個ITAM結構域。在一些實施方案中,膜內結構域中的至少一個包含(CD28+CD3ζ)、(CD28+CD27+CD3ζ)、(CD28+OX40+CD3ζ)、(CD28+4-1BB+CD3ζ)、(CD28+CD27+OX40+CD3ζ)、(CD28+4-1BB+CD27+CD3ζ)、(CD28+4-1BB+OX40+CD3ζ)、(4-1BB+CD3ζ)、(4-1BB+OX40+CD3ζ)、(4-1BB+CD27+CD3ζ)、(CD27+CD3ζ)、(CD27+OX 40+CD3ζ)、(CD28Δ+CD3ζ)、(CD28Δ+CD27+CD3ζ)、(CD28Δ+OX40+CD3ζ)、(CD28Δ+4-1BB+CD3ζ)、(CD28Δ+4-1BB+OX40+CD3ζ)、(CD28Δ+CD27+OX40+CD3ζ)、(CD28Δ+4-1BB+CD27+CD3ζ)、(4-1BB+ICOS+CD3ζ)、(CD28+ICOS+CD3ζ)、(ICOS+CD3ζ)、CD3ζ、或僅CD28。在一些實施方案中,可以測試CAR的活性,例如,使用iQueTM篩選器(iQueTM Screener)(IntelliCyt,Albuquerque,NM)。在一些實施方案中,當在細胞如T細胞中表達時,使用諸如例如流式細胞術等技術,可以評估CAR的一種或多種特征(例如,活力、活化信號的上調、CD25的上調、細胞因子釋放、和/或細胞殺傷)。
本發明的另一個方面涉及一種組合物,所述組合物包含編碼嵌合抗原受體的載體集合,所述載體集合編碼多個獨特的抗原結合結構域、鉸鏈結構域和膜內結構域,所述集合的所述載體就所述結構域而言是隨機化的。
本發明的又一個方面涉及產生多個各自編碼嵌合抗原受體(CAR)的載體的方法,所述方法包括:(i)獲得包含本發明的多個載體(例如,如上文描述)的組合物;并且(ii)使所述組合物經受足以允許在所述載體中包含或由所述載體編碼的所述獨特的抗原結合結構域、鉸鏈結構域和/或膜內結構域經由同源重組而重組的條件以產生多個第四載體,其中所述第四載體中的每個編碼CAR。所述方法可以進一步包括在細胞中表達CAR。所述方法可以進一步包括測試CAR的活性。在一些實施方案中,第一載體中的一個或多個編碼scFv區。在一些實施方案中,第三載體中的一個或多個編碼跨膜結構域。在一些實施方案中,第二載體中的一個或多個編碼跨膜結構域。所述方法可以進一步包括通過重組隨機合并編碼跨膜結構域的第五載體與所述第一載體、第二載體、和第三載體以形成所述第四載體。在一些實施方案中,從編碼多個獨特的scFv區和多個獨特的鉸鏈區的多個載體隨機連接所述第一載體和所述第二載體。在一些實施方案中,從編碼多個獨特的scFv區和多個獨特的膜內結構域的多個載體隨機連接所述第一載體和所述第三載體。在一些實施方案中,從編碼多個獨特的鉸鏈區和多個獨特的膜內結構域的多個載體隨機連接所述第二載體和所述第三載體。在一些實施方案中,從編碼多個獨特的scFv區、多個獨特的鉸鏈區和多個獨特的膜內結構域的多個載體隨機連接所述第一載體、所述第二載體和所述第三載體。所述方法可以進一步包括通過隨機連接來自編碼多個scFv區的載體的第一文庫的所述第一載體,隨機連接來自編碼多個scFv區的載體的第二文庫的所述第二載體,并且隨機連接來自編碼多個膜內結構域的載體的第三文庫的所述第三載體產生所述第四載體,以形成編碼所述CAR的所述第四載體。第一載體可以包含同源重組的第一序列和第二位點。第二載體可以包含同源重組的第二序列和同源重組的第三序列。第三載體可以包含同源重組的所述第三序列和同源重組的第四序列。第三載體可以包含同源重組的所述第三序列和同源重組的第四序列。第四載體可以包含同源重組的所述第一序列和同源重組的所述第四序列。第一載體、第二載體、和/或第三載體可以編碼轉座酶。在一些實施方案中,第六載體編碼轉座酶,并且其中所述方法包括將所述第四載體和所述第六載體中的一種或多種引入、電穿孔、或轉染到細胞中。轉座酶可以是鮭魚型Tc1樣轉座酶(SB)。所述方法可以進一步包括在存在人工抗原呈遞細胞(aAPC)的情況下培養或提供用所述CAR轉染的細胞,所述人工抗原呈遞細胞(aAPC)能刺激表達CAR的T細胞的擴增。在一些實施方案中,所述scFv區、所述鉸鏈區和所述膜內結構域中的每種各自在睡美人(SB)或piggyBac轉座子載體中編碼。在一些實施方案中,從編碼多個獨特的scFv區、鉸鏈區和膜內結構域的多個載體通過所述重組隨機連接所述第一載體、所述第二載體和/或所述第三載體中的每種。在一些實施方案中,所述第一載體、所述第二載體、和所述第三載體各自含有轉座子;并且其中經由同源重組的所述連接包括位點特異性重組。在一些實施方案中,所述第一載體和所述第二載體各自具有第一同源重組位點;并且其中所述第二載體和所述第三載體各自具有第二同源重組位點。在一些實施方案中,所述第一載體具有第三重組位點,并且其中所述第四載體具有第四重組位點,其中所述第三重組位點和第四重組位點可以允許對細胞的同源重組。細胞可以是T細胞,如例如αβT細胞、γδT細胞、或NK細胞、或NKT細胞。在一些實施方案中,細胞是多能細胞,如例如干細胞或誘導的多能干細胞。在一些實施方案中,細胞從干細胞、誘導的多能干細胞或干細胞衍生。細胞可以是從誘導的多能干細胞衍生的T細胞或NK細胞。在一些實施方案中,所述獨特的抗原結合結構域包含至少2、3、4、5、6、7、8、9或更多個選擇性識別不同抗原的scFv。在一些實施方案中,所述獨特的抗原結合結構域包含至少2、3、4、5、6、7、8、9或更多個選擇性識別(即特異性結合)相同抗原的scFv。在一些實施方案中,所述抗原結合結構域選擇性(特異性)結合CD19、通用抗原(小鼠)、HER-3、GD2、Gp75、CS1蛋白、間皮素、磷脂酰絲氨酸、cMyc、CD22、CD4、CD44v6、CD45、CD28、CD3、CD3e、CD123、CD138、CD52、CD56、CD74、CD30、Gp75、CD38、CD33、CD20、Her1/HER3融合物、GD2、碳水化合物、曲霉、ROR1、c-MET、EGFR、Dectin、埃博拉、真菌、GP、HERV-K、NY-ESO-1、VEGF-R2、TGF-b2R、IgG4、生物素或O-AcGD2。在一些實施方案中,所述抗原結合結構域包含scFv或由scFv組成。所述鉸鏈區可以編碼12AA肽(GAGAGCAAGTACGGCCCTCCCTGCCCCCCTTGCCCT,SEQ ID NO:1)、t-20AA肽、IgG4FcΔ EQ、IgG4FcΔQ、(t-12AA+t-20AA)、mKate、phiLov、dsRed、Venus、eGFP、CH3HA,(CD8α+t-20AA)、雙重t-20AA、(t-20AA+CD8α)、(CD8α+亮氨酸拉鏈Basep1)、(CD8α+亮氨酸拉鏈Acid1)、2D3、CD8α或IgG4Fc。膜內結構域可以編碼CD3ζ。膜內結構域可以編碼一個或多個ITAM結構域。在一些實施方案中,所述膜內結構域編碼(CD28+CD3ζ)、(CD28+CD27+CD3ζ)、(CD28+OX40+CD3ζ)、(CD28+4-1BB+CD3ζ)、(CD28+CD27+OX40+CD3ζ)、(CD28+4-1BB+CD27+CD3ζ)、(CD28+4-1BB+OX40+CD3ζ)、(4-1BB+CD3ζ)、(4-1BB+OX40+CD3ζ)、(4-1BB+CD27+CD3ζ)、(CD27+CD3ζ)、(CD27+OX 40+CD3ζ)、(CD28Δ+CD3ζ)、(CD28Δ+CD27+CD3ζ)、(CD28Δ+OX40+CD3ζ)、(CD28Δ+4-1BB+CD3ζ)、(CD28Δ+4-1BB+OX40+CD3ζ)、(CD28Δ+CD27+OX40+CD3ζ)、(CD28Δ+4-1BB+CD27+CD3ζ))、(4-1BB+ICOS+CD3ζ)、(CD28+ICOS+CD3ζ)、(ICOS+CD3ζ)、CD3ζ、或僅CD28。在一些實施方案中,可以測試CAR的活性,例如,使用iQueTM篩選器(IntelliCyt,Albuquerque,NM)。在一些實施方案中,當在細胞如T細胞中表達時,使用諸如例如流式細胞術等技術,可以評估CAR的一種或多種特征(例如,活力、活化信號的上調、CD25的上調、細胞因子釋放、和/或細胞殺傷)。在一些實施方案中,所述活性包括所述CAR選擇性結合癌細胞,選擇性結合病原體,選擇性結合涉及自身免疫性疾病的細胞或者促進T細胞的活化、T細胞的破壞、T細胞的分化、T細胞的增殖、T細胞的去分化、T細胞的移動、T細胞的細胞因子生成、或T細胞的殺傷的能力。
在一些實施方案中,所述癌細胞是卵巢癌、淋巴瘤、腎細胞癌、B細胞惡性腫瘤、CLL、B-ALL、ALL、白血病、B細胞惡性腫瘤或淋巴瘤、套細胞淋巴瘤、無痛性B細胞淋巴瘤、霍奇金淋巴瘤、AML,宮頸癌、乳腺癌、結腸直腸癌、卵巢癌、成神經細胞瘤、皮膚癌、黑素瘤、肺癌、骨肉瘤、膠質瘤、上皮衍生的腫瘤、前列腺癌或兒科癌癥。所述病原體可以是病毒、真菌或細菌。在一些實施方案中,所述測試包括單細胞成像、單細胞遺傳學、單個T細胞或T細胞群體的評估;測量特異性殺傷或連續殺傷、基因表達、蛋白質表達、朝向或遠離靶標的移動、增殖、活化誘導的細胞死亡、細胞因子的分泌或趨化因子的分泌。所述方法可以進一步包括基于單個CAR的特性,從所述多個載體選擇所述單個CAR。所述方法可以進一步包括向受試者治療性施用所述單個CAR。所述受試者可以是哺乳動物,如例如人。
本發明的另一個方面涉及多肽,其包含下列各項或者由下列各項組成:CAR 217(SEQ ID NO:2)、CAR 194(SEQ ID NO:3)、CAR 212(SEQ ID NO:4)、CAR 213(SEQ ID NO:5)、CAR 265(SEQ ID NO:6)、CAR 214(SEQ ID NO:56)、CAR 215(SEQ ID NO:57)、CAR 216(SEQ ID NO:58)、CAR 218(SEQ ID NO:59)、CAR 193(SEQ ID NO:55)或CAR 268(SEQ ID NO:7)。
本發明的又一個方面涉及表達多肽的經轉化的T細胞,所述多肽包含下列各項或者由下列各項組成:CAR 217(SEQ ID NO:2)、CAR 194(SEQ ID NO:3)、CAR 212(SEQ ID NO:4)、CAR 213(SEQ ID NO:5)、CAR 265(SEQ ID NO:6)、CAR 214(SEQ ID NO:56)、CAR 215(SEQ ID NO:57)、CAR 216(SEQ ID NO:58)、CAR 218(SEQ ID NO:59)、CAR 193(SEQ ID NO:55)或CAR 268(SEQ ID NO:7)。細胞可以是永生化細胞。T細胞可以是αβT細胞、γδT細胞、NK細胞、NKT細胞、干細胞、或從干細胞衍生的細胞,包括免疫系統的細胞。
本發明的另一個方面涉及藥物制劑,所述藥物制劑包含本發明的經轉化的T細胞。
本發明的又一個方面涉及核酸,所述核酸編碼包含下列各項或由下列各項組成的嵌合抗原受體:CAR 217(SEQ ID NO:2)、CAR 194(SEQ ID NO:3)、CAR 212(SEQ ID NO:4)、CAR 213(SEQ ID NO:5)、CAR 265(SEQ ID NO:6)、CAR 214(SEQ ID NO:56)、CAR 215(SEQ ID NO:57)、CAR 216(SEQ ID NO:58)、CAR 218(SEQ ID NO:59)、CAR 193(SEQ ID NO:55)或CAR 268(SEQ ID NO:7)。所述核酸可以被包含在T細胞中,如例如αβT細胞、γδT細胞、NK細胞、NKT細胞、干細胞、或從多能細胞衍生的T細胞。T細胞可以被包含在藥學上可接受的載體或賦形劑中。
本發明的另一個方面涉及組合物,所述組合物包含不同CAR編碼載體的文庫,所述文庫的載體就獨特的抗原結合結構域、鉸鏈結構域和/或膜內結構域而言是隨機化的。在一些實施方案中,所述文庫就獨特的抗原結合結構域、鉸鏈結構域和膜內結構域而言隨機化。在一些實施方案中,所述文庫就獨特的抗原結合結構域和膜內結構域而言隨機化。在一些實施方案中,所述文庫就獨特的抗原結合結構域和鉸鏈結構域而言隨機化。在一些實施方案中,所述文庫就獨特的抗原鉸鏈結構域和膜內結構域而言隨機化。
下文在表1中顯示了在本發明的方法中用于產生CAR的抗原結合結構域、鉸鏈區、跨膜結構域和膜內結構域的實例。抗原結合結構域、鉸鏈區、跨膜結構域、和膜內結構域在表1中僅作為非限制性實例提供,并且預期如對于具體臨床應用所期望的,可以選擇實際上任何抗原結合結構域(例如,靶向癌性細胞、細菌、真菌、病毒或病毒感染的細胞)。在表1中,提供了抗原結合結構域的靶標(例如,“CD19”可以指選擇性結合CD19的scFv區)。在一些實施方案中,抗原結合結構域包含選擇性結合抗原的scFv或由選擇性結合抗原的scFv組成。若期望的話,可以在期望時使scFv的部分隨機化(例如,scFv的可變區的部分)。在一些實施方案中,抗原結合結構域選擇性結合蛋白質。或者,抗原結合結構域可以選擇性結合在靶標,如例如真菌、病毒、細菌或癌性細胞上表達的碳水化合物。例如,在一些實施方案中,抗原結合結構域包含Dectin-1或由Dectin-1組成,所述Dectin-1可以選擇性結合存在于真菌細胞壁中的β-葡聚糖和碳水化合物。在一些實施方案中,CAR可以選擇性結合病毒,例如,CAR可以結合病毒蛋白,如肝炎包膜蛋白(例如,Krebs等,2013)。在一些實施方案中,抗原結合結構域是細胞因子。抗原結合結構域可以選擇性結合蛋白質、碳水化合物、或糖。在一些實施方案中,從選擇性結合單一靶標,抗原的多個抗原結合結構域產生CAR,或者抗原結合結構域可以具有重疊的抗原。在一些實施方案中,從選擇性結合不同靶標或抗原的多個抗原結合結構域產生CAR。CAR中的膜內結構域可以在表達CAR的細胞,如例如T細胞或自然殺傷(NK)細胞中產生抑制信號(例如,PD-1、CTLA-4、TIM-3、LAG-3、BTLA、ITIM、SHP-1、LAIR-1、TIGIT、Siglecs)或刺激信號(例如,CD27、CD28、ICOS、CD134、CD137、LCK、DAP10、ITAM、ZAP-70、LAT、SLP-76、細胞因子以及細胞因子受體;以及組合和突變)。當抗原結合區選擇性識別抗原時,膜內結構域可以引起或促進包含CAR的細胞(例如T細胞或NK細胞)活化細胞殺傷、遷移、分化、去分化、或者導致誘導細胞中的凋亡信號。凋亡信號可以包含CTLA4凋亡信號和/或PD1(蛋白質死亡1)凋亡信號或由CTLA4凋亡信號和/或PD1(蛋白質死亡1)凋亡信號組成。在一些實施方案中,可以在細胞,如例如T細胞或NK細胞中表達超過一種獨特的CAR。例如,可以在細胞中表達第一CAR和第二CAR,其中第一CAR選擇性結合健康細胞上的抗原,并且經由第一膜內結構域誘導抑制信號(例如,降低T細胞或NK細胞將損傷健康細胞的概率),并且第二CAR選擇性結合靶細胞(例如,癌性細胞、真菌、病毒感染的細胞、細菌)上的抗原,并且經由第二膜內結構域誘導刺激信號(例如,促進或引起T細胞或NK細胞對靶細胞的殺傷)。經由本發明的方法產生的CAR可以插入靶細胞,如例如T細胞或NK細胞,作為整合的DNA(例如,使用電穿孔和經由轉座酶/轉座子載體或系統的同源重組)或作為非整合的DNA或RNA(例如,使用病毒載體,如例如慢病毒或逆轉錄病毒的mRNA的病毒遞送)。在一些實施方案中,根據本發明的編碼CAR的T細胞是永生化細胞;此類永生化細胞可以發揮功能,可以用于評估或測量CAR的治療潛力或毒性。這樣,可以篩選許多CAR的期望的藥理學譜、針對患病細胞或病原體的毒性、在健康細胞中的毒性缺乏、和/或治療效力。
表1:可以組合為抗原結合結構域-鉸鏈-信號傳導結構域以產生CAR的DNA分子。
ζ–zeta;△-突變體;注意=4-1BB又稱為CD137;“+”指不同區域的融合。
例如,在一些實施方案中,可以使用以下抗原結合結構域、鉸鏈/支架、跨膜結構域、和膜內結構域,如表2中顯示。信號傳導結構域中,例如表1或表2中包含的序列的實例包括CD27(SEQ ID NO:41)、CD28(SEQ ID NO:42)、CD28Δ(SEQ ID NO:43)、CD134(OX40)(SEQ ID NO:44)、CD137(41BB)(SEQ ID NO:45)、ICOS(SEQ ID NO:46)和CD3ζ(SEQ ID NO:47)。如表2中列出的scFv抗EGFR結構域的實例包括Nimotuximab(SEQ ID NO:48)和西妥昔單抗(Cetuximab)(SEQ ID NO:49)。如表2中列出的scFv抗磷脂酰絲氨酸的實例是巴維昔單抗(Bavituximab)(SEQ ID NO:50)。
表2:用于產生CAR的文庫的實例
如本文中使用,術語“嵌合抗原受體(CAR)”或“CAR”包括人工T細胞受體、嵌合T細胞受體、或嵌合免疫受體。CAR一般是工程化受體,其可以將人工特異性植入到特定的免疫效應細胞上。可以采用CAR來將單克隆抗體的特異性賦予給T細胞,從而允許產生大量特異性T細胞,例如用于過繼性細胞療法。在一些實施方案中,CAR將細胞的特異性針對腫瘤相關抗原。在優選的實施方案中,CAR包含膜內結構域(包含胞內活化結構域)、跨膜結構域、鉸鏈或支架區、和包含靶向結構域的胞外結構域(例如,從單克隆抗體衍生的scFv)。在一些實施方案中,胞外靶向結構域可以是受體的配體(例如,選擇性結合蛋白質受體的肽)。在一些實施方案中,可以通過使用對惡性細胞特異的CAR重定向T細胞的特異性(例如,通過使用抗CD19scFv靶向癌性B譜系細胞)來靶向惡性細胞。
表1中顯示了scFv區、鉸鏈/支架區、跨膜結構域、和膜內結構域的實例,并且本文中還提供了相關序列的實例。在表1中注意到scFv區可以指用于特定靶標的多個scFv區(例如,表1中的“CD19”可以指單一單克隆抗體序列,或者在一些優選的實施方案中,它可以指從選擇性靶向CD19的單克隆抗體衍生的多個scFv區)。預期可以使用本發明的方法產生CAR,所述CAR包含例如表1的scFv區、鉸鏈/支架、跨膜結構域、和膜內結構域的任何組合的融合物。例如,在一些實施方案中,CAR可以包含與IgG4Fc鉸鏈/支架區、CD28跨膜結構域、和包含CD28和CD3ζ的膜內結構域融合的選擇性靶向CD19的scFv區(例如,從小鼠、人或人源化單克隆抗體衍生)。在一些實施方案中,CAR可以包含與IgG4Fc鉸鏈/支架區、CD28跨膜結構域、和包含CD28和CD3ζ的膜內結構域融合的選擇性靶向ROR1的scFv區。在一些實施方案中,CAR可以包含與IgG4Fc鉸鏈/支架區、CD28跨膜結構域、和包含4-1BB和CD3ζ的膜內結構域融合的選擇性靶向ROR1的scFv區。在一些實施方案中,CAR可以包含與IgG4Fc鉸鏈/支架區、CD28跨膜結構域、和包含CD28和CD3ζ的膜內結構域融合的選擇性靶向CD19的scFv區(例如,從小鼠、人或人源化單克隆抗體衍生)。
如本文中使用,術語“抗原”是能夠由抗體或T細胞受體結合的分子。抗原一般可以用于誘導體液免疫應答和/或細胞免疫應答,導致B和/或T淋巴細胞的產生。
如本文中說明書使用,“一個”或“一種”可以意指一個/種或多個/種。如本文中在權利要求中使用,當結合詞語“包含”使用時,詞語“一個”或“一種”可以意指一個/種或超過一個/種。
在權利要求書中使用術語“或”用于指“和/或”,除非明確指明僅指備選或者備選是互相排斥的,盡管公開內容支持僅指備選和“和/或”的定義。如本文中使用,“另一”可以意指至少第二或更多。
貫穿本申請,術語“約”用于指數值包括裝置、用于測定數值的方法的固有誤差變化,或者研究受試者間存在的變化。
通過以下詳細描述,本發明的其它目的、特征和優點將變得明顯。然而,應當理解,雖然詳細描述和具體的實例指示本發明的優選的實施方式,但是它們僅作為例示給出,因為通過此詳細描述,在本發明的精神和范圍內的各種變化和修改對于本領域技術人員將變得明顯。
附圖簡述
以下附圖形成本說明書的一部分,并且為了進一步證明本發明的某些方面而包括在內。可以通過與本文中呈現的具體實施方案的詳細描述組合參考這些中的一張或多張圖更好理解本發明。
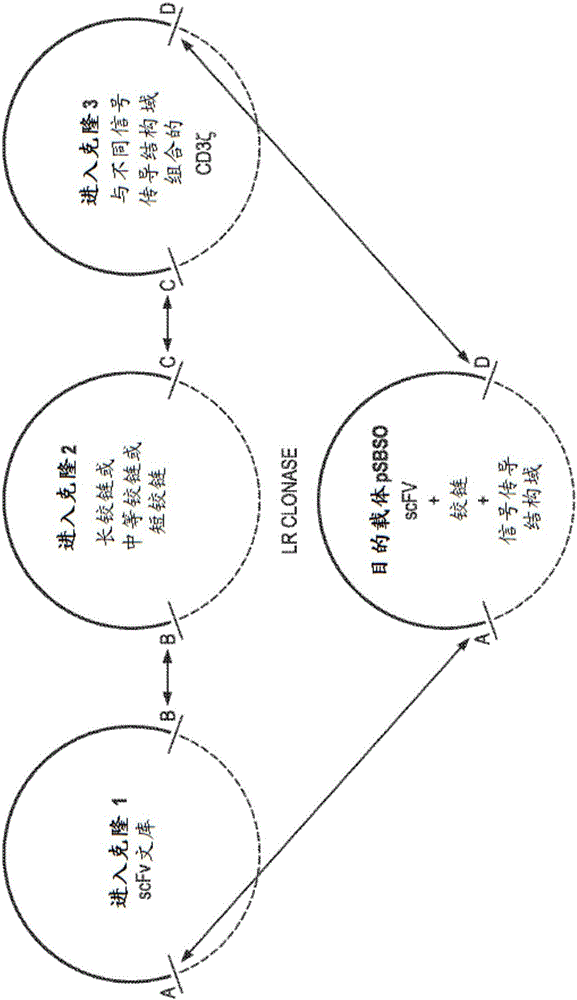
圖1:用于使用表達(i)特異性scFv,(ii)胞外鉸鏈和(iii)膜內結構域的三種供體質粒再裝配CAR的克隆載體。此方法將適合于產生各組在鉸鏈、跨膜和胞內區上不同的CAR。使用三重重組位點系統從組分scFv,IgG4 Fc(長鉸鏈);或CD8a(中等鉸鏈)或僅肽(小鉸鏈)和CD3ζιν與不同信號傳導結構域的組合工程化改造CAR分子。將三種供體質粒(進入克隆)中編碼的scFv的文庫和獨特的支架和信號傳導結構域重組入表達DNA載體中。此方法產生形式scFv-B-支架-C-信號傳導結構域的多個CAR種類。
圖2A-B:(圖2A)通過流式細胞術得到的電穿孔后66天T細胞中的CAR(Fc)和CD8+表達。在加載有CD19抗原的aAPC(克隆4)中擴增細胞。(圖2B)使用4-h鉻釋放測定法通過CD19CAR+T細胞臨床級(CG),通過三重重組位點得到的CD19CAR+T細胞(EZ CAR)和CAR-T細胞將CD19+EL-4的裂解與CD19-EL-4的背景裂解比較。與經照射的且加載有抗CD3(OKT3)的K562衍生的aAPC克隆#4一起擴增CAR-T。
圖3:CAR設計。CAR 212=SEQ ID NO:4;CAR 213=SEQ ID NO:5;CAR 214=SEQ ID NO:56;CAR 215=SEQ ID NO:57;CAR 216=SEQ ID NO:58;CAR 217=SEQ ID NO:2;CAR 218=SEQ ID NO:59;CAR 193=SEQ ID NO:55。
圖4:睡美人追蹤質粒。
圖5:CAR表達。
圖6:CAR表達動力學。
圖7:表型。
圖8A-B:圖8A和圖8B中顯示了延長的表型。
圖9:蛋白質印跡分析。
圖10:擴增動力學。
圖11:倍數擴增:總細胞。
圖12:倍數擴增:CAR+T細胞。
圖13:細胞毒性。
圖14:4-1BB CAR:細胞毒性。
圖15:TM結構域:細胞毒性。
圖16:間隔區(IgG4對CD8):細胞毒性。
圖17:IFN-γ產生。
圖18:4-1BB CAR:IFN-γ產生。
圖19:TM結構域:IFN-γ產生。
圖20:間隔區(IgG4對CD8):IFN-γ產生。
圖21:安全性:針對SB11轉座酶的PCR。
圖22:安全性:CAR拷貝數(qPCR)。
圖23:安全性:自主生長。如圖中顯示,觀察到自主生長的缺乏。
圖24:CAR設計。在圖的右手邊提供了CAR的實例。
圖25:CD3-ζ。詢問=SEQ ID NO:51;對象–頂部=SEQ ID NO:52;對象–中間=SEQ ID NO:53;對象–底部=SEQ ID NO:54。
圖26:CAR設計。
圖27:CAR。
圖28:CAR表達。
圖29:擴增動力學。
圖30:擴增動力學。
圖31:細胞毒性。
圖32:細胞毒性。
圖33:記憶標志物。顯示了CAR+T細胞上的CD27、CD62L、CD28和CCR7的百分比表達(圖26中顯示了表達構建體)。
圖34:IFN-γ產生。
圖35:IFN-γ產生(PMA-Ion)
圖36:自主生長。
圖37:CAR拷貝數。
圖38:CAR拷貝數。
圖39:CAR拷貝數。
圖40A-E:實施通過Lipofectamine用攜帶CAR DNA的質粒(pSBSO EZ CAR)轉染293-HEK細胞。在用抗Fc或抗獨特型(抗CD19svFv)抗體染色后通過流式細胞術分析經轉染的細胞。
圖41A-B:圖41A,Nalm-6;EL-4CD19+細胞;具有MCL和CLL的患者腫瘤細胞(靶標),先前修飾為表達GFP。將5x103個靶細胞與增加濃度的CD19RIgG4CD28CAR T細胞、CD19RCD8αCD28CAR T細胞和CAR-T細胞(用作對照)一起溫育4小時。在4小時后,通過IntelliCyt’s iQue獲得細胞,并且數據分析在其專有的軟件中進行。圖42B,圖代表針對腫瘤細胞的CAR T細胞的殺傷百分比。效應細胞和靶細胞之前的比率范圍為0至40個細胞。
圖42:將5x103個靶細胞(EL-4CD19+粒酶B細胞報告物)與增加濃度的臨床級CD19RIgG4CD28CAR T細胞、EZ CD19RCD8α CD28 CAR T細胞和CAR-T細胞(用作對照)一起溫育4小時和10小時。在溫育時間后,通過IntelliCyt’s iQue獲得細胞,并且數據分析在其專有的軟件中進行。圖代表針對腫瘤細胞的CAR T細胞的殺傷百分比。效應細胞和靶細胞之前的比率范圍為0至20個細胞。
具體實施方式
本文中提供了用于產生嵌合抗原受體(CAR)的方法。所述方法利用多個載體,每個編碼抗原結合結構域(例如,scFv區)、鉸鏈區、跨膜區、和/或膜內結構域。例如,在一些實施方案中,第一載體編碼抗原結合結構域,第二載體編碼鉸鏈區,并且第三區編碼膜內結構域。在一些實施方案中,跨膜區在第二載體中,在第三載體中,或在第四載體中編碼。在一些優選的實施方案中,載體可以同源重組以形成編碼CAR的核酸,所述CAR包含抗原結合結構域、鉸鏈區、跨膜區、和膜內結構域。這樣,可以產生許多CAR,并且篩選期望的活性,如例如,選擇性識別和殺傷表達由CAR選擇性結合的抗原的癌性細胞。然后,CAR可以在細胞,如T細胞或自然殺傷(NK)細胞中以整合核酸(例如,使用轉座酶/轉座子整合到宿主基因組中的DNA)或以非整合核酸(例如,經由病毒載體,如慢病毒或逆轉錄病毒遞送的mRNA)表達。然后,表達CAR的T細胞或NK細胞可以在藥物制劑或賦形劑中向受試者,如人患者施用以治療或預防疾病(例如,癌癥、真菌感染、細菌感染、或病毒感染)。
I.文庫產生
可以通過本領域技術人員已知的方法產生編碼多個scFv區、鉸鏈/支架區、跨膜結構域、和膜內結構域(信號傳導結構域)的文庫。在一些實施方案中,對于scFv區、鉸鏈/支架區、和膜內結構域(信號傳導結構域)中的兩個或三個,可得到多種可能性。在一些實施方案中,對于scFv區、鉸鏈/支架區、跨膜結構域、和膜內結構域(信號傳導結構域)中的兩個、三個或全部,可得到多種可能性。例如,在表1中提供了scFv區、鉸鏈/支架區、跨膜結構域、和膜內結構域(信號傳導結構域)的實例。在一些實施方案中,文庫可以編碼多個靶向不同抗原,如多個抗癌抗原或腫瘤靶向抗原的scFv;在其它實施方案中,文庫可以編碼選擇性結合單一靶標(例如,單一抗癌抗原,如CD19等)的多個不同scFv。這樣,可以使用所述方法鑒定哪個腫瘤靶向構建體可以對給定細胞樣品更有效起(例如以在個體化藥物中使用)或可以使用所述方法鑒定在靶向給定抗原中更有效發揮功能的新CAR。scFv區一般包含從抗體衍生的可變輕(VL)和可變重(VH)鏈。在一些實施方案中,若想要的話,可以隨機化VL和VH區的部分。用于產生文庫的通用方法包括例如產生酵母文庫、細菌、噬菌體文庫、浸潤性B細胞、雜交瘤(包括來自人和嚙齒類),或來自羊駝(llamas)的文庫、駱駝、馬文庫、和計算機方法(參見例如Lennard,2002)。
在一些實施方案中,融合編碼scFv、鉸鏈/支架區、跨膜結構域、和膜內結構域的不同載體以形成編碼CAR的單一載體。可以經由轉座子介導的同源重組發生融合。
例如,在一些實施方案中,編碼scFv、鉸鏈/支架區、跨膜結構域、和/或膜內結構域的載體可以是睡美人(SB)或piggyBac DNA質粒。睡美人(SB)和piggyBac DNA質粒描述于例如Maiti等(2013),Singh等(2008),和Huls等(2013)。在一些實施方案中,轉座子由鮭魚型Tc1樣轉座酶(SB)介導。在一些優選的實施方案中,經由如Singh等,2014或Huls等中描述的方法,將編碼CAR的載體轉染或摻入來自受試者,如患有癌癥的人患者的T細胞中。例如,可以使用從睡美人(SB)系統衍生的DNA載體避免與用重組病毒載體轉導T細胞有關的費用和制造困難。在電穿孔后,轉座子/轉座酶可以改善用于在T細胞中表達CAR和其它轉基因的質粒的整合效率。與人工抗原呈遞細胞(aAPC)組合的SB系統可以選擇性增殖,并且產生適合于人應用的CAR(+)T細胞。在一些實施方案中,兩種DNA質粒、SB轉座子(編碼感興趣的CAR)和SB轉座酶(例如,SB11)的同步電轉移可以繼之以通過在存在可溶性重組人IL-2和IL-21的情況下每7天添加(刺激循環)經γ-照射的aAPC回收穩定整合體。例如,可以進行4個循環(28天的連續培養)以產生臨床上有吸引力的數目的穩定表達感興趣CAR的T細胞。可以利用轉座子/轉座酶系統的使用遞送表達CAR的T細胞,如例如Hackett等中描述。
II.嵌合抗原受體
本發明的實施方案牽涉產生并鑒定編碼抗原特異性嵌合抗原受體(CAR)多肽的核酸。在一些實施方案中,將CAR進行人源化以降低免疫原性(hCAR)。
在一些實施方案中,CAR可以識別由一種或多種抗原之間共享的空間構成的表位。模式識別受體,如Dectin-1可以用于驅動針對碳水化合物抗原的特異性。在某些實施方案中,結合區可以包含單克隆抗體的互補決定區、單克隆抗體的可變區、和/或其抗原結合片段。在一些實施方案中,結合區是scFv。在另一個實施方案中,結合受體或細胞靶標的肽(例如,細胞因子)可以作為可能性包括在內或者替換CAR的結合區中的scFv區。因此,在一些實施方案中,可以從編碼多個scFv區和/或其它靶向蛋白的多個載體產生CAR。互補決定區(CDR)是在抗原受體(例如,免疫球蛋白和T細胞受體)蛋白的可變域中找到的互補抗原并且因此向受體提供其針對所述特定抗原的特異性的短氨基酸序列。抗原受體的每條多肽鏈含有三個CDR(CDR1、CDR2和CDR3)。由于抗原受體通常由兩條多肽鏈構成,每個抗原受體有可以與抗原接觸的6個CDR:每條重鏈和輕鏈含有3個CDR。由于通常在CDR中找到與免疫球蛋白和T細胞受體選擇性有關的大多數序列變異,這些區域有時稱為超變結構域。在這些之中,CDR3顯示最大的變異性,因為它由VJ(在重鏈和TCR αβ鏈的情況下為VDJ)區的重組編碼。
經由本發明產生的CAR編碼核酸可以包含一個或多個人基因或基因片段以增強用于人患者的細胞免疫療法。在一些實施方案中,可以經由本文中描述的方法產生全長CAR cDNA或編碼區。抗原結合區或結構域可以包含從特定的人單克隆抗體衍生的單鏈可變片段(scFv)的VH和VL鏈的片段,如那些描述于美國專利7,109,304(通過引用方式并入本文)的片段。在一些實施方案中,scFv包含人抗原特異性抗體的抗原結合結構域。在一些實施方案中,scFv區是由針對人密碼子使用優化以在人細胞中表達的序列編碼的抗原特異性scFv。
CAR的抗原結合結構域的排列可以是多聚體的,如雙抗體(diabody)或多聚體。多聚體可以通過將輕鏈和重鏈的可變部分交叉配對成可以稱為雙抗體的事物而形成。在一些實施方案中,可以縮短或排除CAR的鉸鏈部分(即,產生僅包含抗原結合結構域、跨膜區和胞內信號傳導結構域的CAR)。大量鉸鏈可以在本發明的情況下使用,例如如表1中顯示。在一些實施方案中,可以使鉸鏈區的第一個半胱氨酸得到維持,或者通過脯氨酸或絲氨酸取代突變,或者截短直至第一個半胱氨酸。從用作抗原結合區的scFv缺失Fc部分以產生根據本發明的CAR。在一些實施方案中,抗原結合區可以編碼僅Fc結構域之一,例如來自人免疫球蛋白的CH2或CH3結構域。還可以包括已經被修飾以改善二聚化和寡聚化的人免疫球蛋白的鉸鏈、CH2、和CH3區。在一些實施方案中,鉸鏈部分可以包含8-14個氨基酸的肽(例如12AA肽)、CD8α的部分、或IgG4Fc或者由其組成。在一些實施方案中,可以使用促進寡聚化的結構域,如CD8α,從細胞表面附加抗原結合結構域。在一些實施方案中,可以使用被單克隆抗體(mAb)克隆2D3(描述于例如Singh等,2008中的mAb克隆2D3)識別的結構域從細胞表面附加抗原結合結構域。
CAR的膜內結構域或胞內信號傳導結構域一般可以引起或促進包含CAR的免疫細胞的至少一種正常效應子功能的活化。例如,膜內結構域可以促進T細胞的效應子功能,如例如細胞裂解活性或輔助活性,包括細胞因子的分泌。初始、記憶的或記憶類型的T細胞中的效應子功能可以包括抗原依賴性增殖。術語“胞內信號傳導結構域”或“膜內結構域”指可以轉導效應子功能信號和/或指導細胞實施特化功能的CAR部分。雖然通常整個胞內信號傳導結構域可以被包含在CAR中,但是在一些情況中可以包含膜內結構域的截短部分。一般地,膜內結構域包括截短的膜內結構域,其中截短的膜內結構域保留在細胞中轉導效應子功能信號的能力。
在一些實施方案中,膜內結構域包含T細胞受體的ζ鏈或任何其同源物(例如,η、δ、γ、或ε)、MB1鏈、B29、Fc RIII、Fc RI和信號傳導分子的組合,如CD3ζ和CD28,CD27、4-1BB、DAP-10、OX40及其組合,以及其它類似的分子和片段。可以使用活化蛋白家族的其它成員的胞內信號傳導部分,如FcγRIII和FcεRI。這些備選的跨膜和胞內結構域的實例可以參見例如Gross等(1992),Stancovski等(1993),Moritz等(1994),Hwu等(1995),Weijtens等(1996),和Hekele等(1996),通過引用方式以其整體并入本文。在一些實施方案中,膜內結構域可以包括人CD3ζ胞內域。
優選地,通過跨膜結構域連接抗原特異性胞外結構域和胞內信號傳導結構域。可以在CAR中包含的跨膜結構域包括例如人IgG4Fc鉸鏈和Fc區、人CD4跨膜結構域、人CD28跨膜結構域、跨膜人CD3ζ結構域、或半胱氨酸突變人CD3ζ結構域、或來自人跨膜信號傳導蛋白如例如CD16和CD8和紅細胞生成素受體的跨膜結構域。例如,在表1中提供了跨膜結構域的實例。
在一些實施方案中,膜內結構域包含編碼共刺激受體,如例如經修飾的CD28胞內信號傳導結構域、或CD28、CD27、OX-40(CD134)、DAP10、或4-1BB(CD137)共刺激受體的序列。在一些實施方案中,由CD3ζ啟動的初始信號,由人共刺激受體提供的別的信號兩者可以被包含在CAR中以更有效活化經轉化的T細胞,這可以有助于改善過繼性免疫療法的體內持久性和治療成功。如表1中記錄,膜內結構域或胞內受體信號傳導結構域可以包含單獨或與FcγRIII共刺激信號傳導結構域,如例如CD28、CD27、DAP10、CD137、OX40、CD2、4-1BB組合的CD3的ζ鏈。在一些實施方案中,膜內結構域包含TCRζ鏈、CD28、CD27、OX40/CD134、4-1BB/CD137、FcεRIγ、ICOS/CD278、IL-2Rbeta/CD122、IL-2Ralpha/CD132、DAP10、DAP12、和CD40中的一種或多種的部分或全部。在一些實施方案中,膜內結構域中可以包含1、2、3、4或更多個胞質結構域。例如,在一些CAR中,已經觀察到融合在一起的至少兩個或三個信號傳導結構域可以導致疊加或協同效應。
在一些方面,可以產生包含編碼CAR的DNA序列的分離的核酸區段和表達盒。可以使用多種載體。在一些優選的實施方案中,載體可以允許將編碼CAR的DNA遞送到免疫,如T細胞。CAR表達可以在調控的真核啟動子,如例如MNDU3啟動子、CMV啟動子、EF1α啟動子、或泛素啟動子的控制下。此外,載體可以含有選擇標記,若不是出于其它原因,以促進其體外操作。在一些實施方案中,可以從自DNA模板體外轉錄的mRNA表達CAR。
嵌合抗原受體分子是重組的,并且以其既結合抗原又經由存在于其胞質尾部中的免疫受體活化基序(ITAM)而轉導活化信號的能力區分。利用抗原結合部分(例如,從單鏈抗體(scFv)產生)的受體構建體提供“通用”的額外優點,因為它們可以以不依賴于HLA的方式結合靶細胞上的天然抗原。例如,可以將scFv構建體與編碼CD3復合物的ζ鏈(ζ)的胞內部分、Fc受體γ鏈、和sky酪氨酸激酶的序列融合(Eshhar等,1993;Fitzer-Attas等,1998)。已經在幾種鼠和人抗原-scFv:ζ系統中證明重定向的T細胞效應子機制,包括CTL的腫瘤識別和裂解(Eshhar等,1997;Altenschmidt等,1997;Brocker等,1998)。
例如,抗原結合區可以來自人或非人scFv。使用非人抗原結合區,如鼠單克隆抗體的一個可能的問題是降低的人效應子功能性和降低的穿透腫瘤塊的能力。此外,非人單克隆抗體可以被人宿主識別為外來蛋白質,并且因此重復注射此類外來抗體可以導致免疫應答的誘導,從而導致有害的超敏感性反應。對于基于鼠的單克隆抗體,此效應已經稱為人抗小鼠抗體(HAMA)應答。在一些實施方案中,與一些鼠抗體相比,在CAR中包含人抗體或scFv序列可以導致很少或無HAMA應答。類似地,在CAR中包含人序列可以用于降低或避免免疫介導的內源T細胞的識別或消除的風險,所述內源T細胞存在于接受者中,并且可以基于HLA識別加工過的抗原。
在一些實施方案中,CAR包含:a)胞內信號傳導結構域,b)跨膜結構域,c)鉸鏈區,和d)包含抗原結合區的胞外結構域。在一些實施方案中,胞內信號傳導結構域和跨膜結構域與膜內結構域一起由單一載體編碼,所述單一載體可以與編碼鉸鏈區的載體和編碼抗原結合區的載體融合(例如,經由轉座子指導的同源重組)。在其它實施方案中,胞內信號傳導區和跨膜區可以由融合(例如,經由轉座子指導的同源重組)的兩個不同載體編碼。
在一些實施方案中,CAR的抗原特異性部分,又稱為包含抗原結合區的胞外結構域,選擇性靶向腫瘤相關抗原。腫瘤相關抗原可以是任何種類的,只要它在腫瘤細胞的細胞表面上表達。可以用經由本發明產生的CAR靶向的腫瘤相關抗原的實例包括例如CD19、CD20、癌胚抗原、甲胎蛋白、CA-125、MUC-1、CD56、EGFR、c-Met、AKT、Her2、Her3、上皮腫瘤抗原、黑素瘤相關抗原、突變p53、突變ras、Dectin-1等。在一些實施方案中,CAR的抗原特異性部分是scFv。表1中提供了腫瘤靶向性scFv的實例。在一些實施方案中,CAR可以與膜結合細胞因子共表達,例如以改善在存在有少量腫瘤相關抗原時的持久性。例如,CAR可以與膜結合IL-15共表達。
在一些實施方案中,可以用CAR靶向胞內腫瘤相關抗原,如例如HA-1、存活蛋白、WT1和p53。這可以通過通用T細胞上表達的CAR實現,所述通用T細胞在HLA的背景中識別從胞內腫瘤相關抗原描述的加工過的肽。另外,通用T細胞可以遺傳修飾為表達T細胞受體配對,其在HLA的情形中識別胞內加工過的腫瘤相關抗原。
由CAR識別的病原體可以基本上是任何種類的病原體,但是在一些實施方案中,病原體是真菌、細菌或病毒。示例性的病毒病原體包括腺病毒科(Adenoviridae)、埃巴病毒(EBV)、巨細胞病毒(CMV)、呼吸道合胞病毒(RSV)、JC病毒、BK病毒、HSV、HHV病毒科、細小RNA病毒科(Picornaviridae)、皰疹病毒科(Herpesviridae)、嗜肝DNA病毒科(Hepadnaviridae)、黃病毒科(Flaviviridae)、逆轉錄病毒科(Retroviridae)、正粘病毒科(Orthomyxoviridae)、副粘液病毒科(Paramyxoviridae)、乳多空病毒科(Papovaviridae)、多瘤病毒(Polyomavirus)、彈狀病毒科(Rhabdoviridae)、和披膜病毒科(Togaviridae)的病毒。示例性的病原性病毒引起天花、流感、流行性腮腺炎、麻疹、水痘、埃博拉和風疹。示例性的病原性真菌包括念珠菌屬(Candida)、曲霉屬(Aspergillus)、隱球菌屬(Cryptococcus)、組織胞漿菌屬(Histoplasma)、肺囊蟲屬(Pneumocystis)和葡萄狀穗霉屬(Stachybotrys)。示例性的病原性細菌包括鏈球菌屬(Streptococcus)、假單胞菌屬(Pseudomonas)、志賀菌屬(Shigella)、彎曲桿菌屬(Campylobacter)、葡萄球菌屬(Staphylococcus)、螺桿菌屬(Helicobacter)、大腸桿菌(E.coli)、立克次體屬(Rickettsia)、芽孢桿菌屬(Bacillus)、博德特菌屬(Bordetella)、衣原體屬(Chlamydia)、螺旋體屬(Spirochetes)、和沙門氏菌屬(Salmonella)。在一些實施方案中,病原體受體Dectin-1可以用于產生CAR,所述CAR識別真菌,如曲霉屬的細胞壁上的碳水化合物結構。在另一個實施方案中,可以基于識別病毒決定簇(例如,來自CMV和埃博拉的糖蛋白)的抗體制備CAR以中斷病毒感染和病理學。
在一些實施方案中,可以將編碼CAR的裸DNA或合適的載體引入受試者的T細胞(例如,從患有癌癥或其它疾病的人患者獲得的T細胞)中。通過使用裸DNA的電穿孔穩定轉染T細胞的方法是本領域中已知的。參見例如美國專利No.6,410,319。裸DNA一般指以對于表達正確的方向在質粒表達載體中含有的編碼本發明的嵌合受體的DNA。在一些實施方案中,裸DNA的使用可以減少產生表達經由本發明的方法產生的CAR的T細胞需要的時間。
或者,病毒載體(例如,逆轉錄病毒載體、腺病毒載體、腺相關病毒載體、或慢病毒載體)可以用于將嵌合構建體引入T細胞中。一般地,用于轉染來自受試者的T細胞的編碼CAR的載體在受試者的T細胞中一般應當是非復制的。基于病毒的大量載體是已知的,其中在細胞中維持的病毒的拷貝數足夠低以維持細胞的活力。說明性的載體包括pFB-neo載體以及基于HIV、SV40、EBV、HSV或BPV的載體。
一旦確定經轉染或轉導的T細胞能夠以期望的調節和在期望水平以表面膜蛋白表達CAR,可以確定嵌合受體在宿主細胞中是否有功能以提供期望的信號誘導。隨后,可以向受試者再引入或施用經轉導的T細胞以活化受試者中的抗腫瘤應答。為了促進施用,可以用合適的載體或稀釋劑(其優選是藥學上可接受的)將經轉導的T細胞制備成藥物組合物或者制備成適合于體內施用的植入物。本領域中已經描述了制備此類組合物或植入物的手段(參見例如Remington's Pharmaceutical Sciences,第16版,Mack編(1980))。在適當時,可以將表達CAR的經轉導的T細胞配制成半固體或液體形式的制劑,如膠囊、溶液、注射液、吸入劑、或氣霧劑,以用于其各自的施用途徑的常見方式進行。可以利用本領域中已知的手段防止或最小化組合物的釋放和吸收,直至它達到靶組織或器官,或者確保組合物的定時釋放。一般地,優選地,采用不使表達嵌合受體的細胞失效(ineffectuate)的藥學可接受形式。因此,期望地,可以將經轉導的T細胞制備成含有平衡鹽溶液,如漢克(Hanks)氏平衡鹽溶液、或生理鹽水的藥物組合物。
IV.人工抗原呈遞細胞
在一些情況中,aAPC可用于制備基于CAR的治療組合物和細胞療法產品。對于關于抗原呈遞系統的制備和用途的一般指導,參見例如美國專利No.6,225,042、6,355,479、6,362,001和6,790,662;美國專利申請公開No.2009/0017000和2009/0004142;和國際公開No.WO2007/103009)。
可以使用aAPC擴增表達CAR的T細胞。在遇到腫瘤抗原期間,通過抗原呈遞細胞對T細胞遞送的信號可以影響T細胞編程及其隨后的治療效力。這已經刺激開發人工抗原呈遞細胞的努力,所述人工抗原呈遞細胞允許最佳控制對T細胞提供的信號(Turtle等,2010)。除了感興趣的抗體或抗原外,aAPC系統還可以包含至少一種外源輔助分子。可以采用輔助分子的任何合適的數目和組合。輔助分子可以選自諸如共刺激分子和粘附分子等輔助分子。示例性的共刺激分子包括CD70和B7.1(又稱作B7或CD80),其可以結合T細胞表面上的CD28和/或CTLA-4分子,從而影響例如T細胞擴增、Th1分化、短期T細胞存活、和細胞因子分泌,如白介素(IL)-2(參見Kim等,2004)。粘附分子可以包括碳水化合物結合糖蛋白,如選擇蛋白、跨膜結合糖蛋白,如整聯蛋白、鈣依賴性蛋白,如鈣粘蛋白和單次跨膜免疫球蛋白(Ig)超家族蛋白質,如胞間粘附分子(ICAM),其促進例如細胞與細胞或細胞與基質接觸。示例性的粘附分子包括LFA-3和ICAM,如ICAM-1。例如,在美國專利No.6,225,042、6,355,479和6,362,001中例示了可用于選擇、克隆、制備和表達示例性的輔助分子,包括共刺激分子和粘附分子的技術、方法和試劑。
優選地,為了變為aAPC而選擇的細胞具有胞內抗原呈遞、胞內肽運輸、和/或胞內MHC I類或II類分子-肽加載的缺陷,或者是變溫的(即,比哺乳動物細胞系對溫度攻擊的敏感性更小),或者擁有缺陷和變溫特性兩者。優選地,為了變為aAPC而選擇的細胞還缺乏表達引入細胞中的外源MHC I類或II類分子和輔助分子組分的至少一種內源對應物(例如,如上文描述的內源MHC I類或II類分子和/或內源輔助分子)的能力。此外,aAPC優選地保留細胞在其修飾為產生aAPC前擁有的缺陷和變溫特性。示例性的aAPC構成或衍生自與抗原加工(TAP)缺陷細胞系,如昆蟲細胞系有關的轉運蛋白。示例性的變溫昆蟲細胞系是果蠅細胞系,如Schneider 2細胞系(例如Schneider,J.m 1972)。美國專利No.6,225,042、6,355,479和6,362,001中提供了用于Schneider 2細胞的制備、生長和培養的說明性方法。
aAPC可以進行冷凍-融化循環。例如,可以通過將含有aAPC的合適容器與合適量的液氮、固體二氧化碳(干冰)、或類似的低溫材料接觸,從而快速發生冷凍來冷凍aAPC。然后,通過從低溫材料取出aAPC并且暴露于周圍室溫條件,或者通過其中采用微溫水浴或溫手促進較短的融化時間的促進融化過程融化冷凍的aAPC。另外,可以冷凍aAPC,并且在融化前貯存延長的時間段。也可以融化冷凍的aAPC,然后在進一步使用前凍干。有利地,可以不利影響冷凍-融化程序的防腐劑,如二甲亞砜(DMSO)、聚乙二醇(PEG)和其它防腐劑可以從進行冷凍-融化循環的含有aAPC的培養基中缺乏,或者基本上被除去,如通過將aAPC轉移到基本上缺乏此類防腐劑的培養基。
在其它優選的實施方案中,可以通過交聯使異體核酸和aAPC的內源核酸失活,從而在失活后基本上不發生細胞生長、核酸的復制或表達。例如,可以在表達外源MHC和輔助分子,在aAPC表面上呈遞此類分子,并且給呈遞的MHC分子加載一種或多種選擇的肽后的點時使aAPC失活。因而,雖然此類加載有失活的且選擇的肽的aAPC使得基本上不能增殖或復制,但是它們可以保留選擇的肽呈遞功能。交聯也可以導致基本上沒有污染性微生物,如細菌和病毒的aAPC,而基本上不降低aAPC的抗原呈遞細胞功能。因此,交聯可以用于維持aAPC的重要APC功能,同時幫助減輕關于使用aAPC開發的細胞療法產品的安全性的憂慮。關于與交聯和aAPC相關的方法,參見例如美國專利申請公開No.20090017000,其通過引用方式并入本文。
IV.實施例
包括以下實施例以證明本發明的優選的實施方案。本領域技術人員應當理解,以下實施例中公開的技術代表由發明人發現的在本發明的實施中良好發揮功能的技術,并且因此可以認為構成其優選的實施模式。然而,根據本公開內容,本領域技術人員應當理解可以在不偏離本發明的精神和范圍的前提下對公開的具體的實施方案進行許多改變,而仍然獲得相同或相似的結果。
實施例1
材料和方法
臨床級DNA質粒的產生
SB轉座子CoOpCD19RCD28ζ/pSBSO在EF-1/HTLV雜合復合啟動子(InvivoGen)下表達人密碼子優化的(CoOp)第二代CoOpCD19RCD28ζCAR,所述EF-1/HTLV雜合復合啟動子(InvivoGen)由延長因子-1a(EF-1a[Kim等,1990]和人T細胞白血病病毒(HTLV)的59非翻譯區[Singh等,2011;Davies等,2010]構成。此DNA質粒的衍生描述于圖S1中。從DNA質粒pCMV-SB11以順式表達在巨細胞病毒(CMV)啟動子下的SB轉座酶SB11(Singh等,2011;Singh等,2008)。對這兩種質粒完整測序,并且由Waisman Clinical Biomanufacturing Facility(Madison,WI)使用用于選擇細菌菌株大腸桿菌DH5a的卡那霉素制備。
產生三重位點特異性重組DNA質粒:EZ-Build-CAR
使用來自上文描述的CAR(CoOpCD19RCD28z/pSBSO)的DNA序列,各部分CD19ScFv、鉸鏈IgG4Fc和與CD3ζ信號傳導結構域偶聯的域結構域CD28跨膜和胞質溶膠部分側翼有λ重組位點,由Geneart(Life Technologies)以PCR產物合成。將這三個部分單獨插入pDonors221質粒中(通過酶BP clonase(兩者都來自Invitrogen)。將三個質粒與三重位點特異性重組睡美人質粒通過酶LR PLUS clonase(Invitrogen)重組,以scFv-B-支架-C-信號傳導結構域形式產生EZ-Build CD19CD28ζCAR(圖1)。
細胞計數
使用臺盼藍排除法(Trypan-blue exclusion)區分活細胞與死亡細胞,并且使用Cellometer(Nexecelom Bioscience)計數(Singh等,2011)。
PBMC的分離
來自兩名男性志愿者健康供體的白細胞去除術(Leukapheresis)產品購自Key Biologics LLC(Memphis,TN)。通過我們修改Biosafe Sepax系統(Eysins,Switzerland)根據cGMP工作分離外周血單個核細胞(PBMC)。簡言之,在閉合CS-900試劑盒上的所有夾子后,經由60mL注射器通過魯爾鎖緊連接器(Luer-lock connector)將100mL Ficoll(GE Healthcare)無菌轉移到密度梯度介質袋(“ficoll袋”),并且加熱使用手持式密封器(Sebra,型號#2380)密封的管道。將試劑盒用釘連接到1,000mL含有具有20mL 25%人血清清蛋白(HSA)(Baxter)的CliniMACS緩沖液(PBS/EDTA,Miltenyi,Cat#70026)(2%v/v,清洗緩沖液)以用于清洗的袋、最終產品袋[300mL具有偶聯器的轉移包(Baxter/Fenwal 4R2014)]和試劑/血液袋。使用基于密度梯度的分離方案(v126),將注射器活塞加載到離心機室中,并且將Sepax蓋加載到aAPC(克隆#4)中以選擇性增殖CAR+T細胞。使用經c-照射的aAPC在數字上擴增經遺傳修飾的T細胞。在VueLife細胞培養袋中在CM中使來自WCB的融化的aAPC增殖長達60天,并且使用Biosafe Sepax II收獲程序收獲。簡言之,經由魯爾鎖定連接將CS-490.1試劑盒連接到300mL輸出袋(轉移包)。在坑中安裝分離室,并且將管道插入光學傳感器中,并且旋塞閥對準成T位置。在連接壓力傳感器線后,在支持架上吊起產品袋和上清液/血漿袋。修改的方案PBSCv302選自Sepax手冊,并且有待處理的輸入產品的體積(初始體積)設置為#840mL。在確認和試劑盒測試后,開始程序。在完成后,除去袋,閉合夾子,并且除去試劑盒。無菌取出來自最終產品袋的細胞,用清洗介質(Plasmalyte中的10%HSA)清洗兩次,并且計數。使用CIS BIO國際放射器(IBL-437C#09433)照射(100Gy)aAPC,并且使用控制速率冷凍器(Planer Kryo 750)在冷凍保存介質中冷凍保存備用。使用加載有抗CD3(OKT3)的K562衍生的aAPC克隆#4來增殖尚未進行遺傳修飾的對照(CAR-)自體對照T細胞。在稱作加載培養基(LM)的含有0.2%乙酰半胱氨酸(Acetadote,Cumberland Pharmaceuticals)的無血清X-Vivo 15(目錄號04-744Q,Lonza)中將獲自培養物的aAPC溫育過夜。次日,洗滌細胞,使用γCell 1000 Elite Cs-137放射器(MDS Nordion)照射(100Gy),與1mg/106細胞功能級純化的抗人CD3(克隆-OKT3,16-0037-85,eBioscience)一起以106個細胞/mL的濃度重懸于LM中,并且在3-D旋轉器(Lab-Line)上在溫和攪拌的情況下于4℃溫育30分鐘。在用LM洗滌三次后,細胞用于實驗或者在氣體層中在液氮中以等分試樣冷凍以備用。
CAR+T細胞的制備
將融化的PBMC重懸于(i)人T細胞試劑盒(目錄號VPA-1002,Lonza;對于2x107個細胞,一個小杯中100μL)中,與(ii)編碼CD19RCD28CAR轉座子的DNA質粒(CoOpCD19RCD28/pSBSO)(每個小杯每2x107個PBMC的15μg超螺旋DNA),和(iii)編碼SB11轉座酶的DNA質粒(pCMVSB11)(每個小杯每2x107個PBMC的5μg超螺旋DNA)。將此混合物立即轉移到小杯(Lonza),使用Nucleofector II(程序U-14,Amaxa/Lonza)電穿孔(限定培養第0天),在10%RPMI完全培養基中靜止2至3小時,并且在半培養基更換后,在37℃,5%CO2溫育過夜。次日,收獲細胞,計數,通過流式細胞術測定表型,并且以比率1:2(CAR+T細胞:aAPC)與經c-照射的aAPC共培養,其標記為培養第1天和7天刺激周期的開始。分別從第1天和第7天起按星期一-星期三-星期五日程表添加IL-21(目錄號AF-200-21,PeproTech)和IL-2(目錄號NDC 65483-116-07,Novartis)。NK細胞可以防止CAR+T細胞的數字擴增,尤其在它們的過度生長在組織培養過程中早期出現的情況下。因此,若CD3-CD56+細胞$10%,則在含有25%HAS的CliniMACS緩沖液(80mL/107個細胞)中在LS柱(目錄號130-042-401,Miltenyi Biotech)上使用CD56珠粒(目錄號70206,Miltenyi Biotech,20mL珠/107個細胞)實施CD56消減。
CAR-對照T細胞的產生
作為對照,在7天刺激循環中以1:1的比率將5x106個mock轉染的PBMC與經照射的并且加載有抗CD3(OKT3)的K562衍生的aAPC克隆#4共培養。從培養第1天起給所有培養物補充IL-21(30ng/mL),并且在開始培養后7天開始補充IL-2(50U/mL)。隨后,每隔一天添加所有細胞因子。
細胞的免疫表型
在4℃使用100mL FACS緩沖液(2%FBS,0.1%疊氮化鈉)中的抗體使染色細胞30分鐘。使用FACSCalibur(BD Bioscience)進行采集,并且使用FCS Express 3.00.0612分析。
鉻釋放測定
使用經51Cr標記的靶細胞,在標準的4小時鉻釋放測定中對T細胞評估其細胞毒性。一式三份在1x105、0.5x105、0.25x105、0.125x105底部板(Costar)上鋪板T細胞。在溫育后,將50μL上清液收獲到LumaPlate(Perkin Elmer)上,在TopCount NXT(Perkin Elmer)上讀數,并且如下計算特異性裂解百分比:
實驗51Cr釋放-自發51Cr釋放 x 100
最大51Cr釋放-自發51Cr釋放
通過在96孔V(Manufacturee Novo Software,Thornhill,Ontario,Canada)中在來自分別與CM或0.1%Triton X-100(Sigma)溫育的靶細胞和0.0625x105個細胞/孔與5x103個靶細胞的條件化上清液中測量鉻來測定自發和最大釋放。
實施例2
CD19+CAR的產生
使用上文在實施例1中描述的方法(稱為“EZ”法)產生CD19+CAR。比較這些CD19+CAR(CD19CAR)與經由先前的方法產生的臨床級(“CG”)CD19CAR。
數據顯示了三重位點重組系統產生與臨床級CD19CAR(CG)相似的CD19CAR(EZ)。質粒中由重組位點留下的足跡不干擾CAR的表達和功能(圖2A-B)。
實施例3
含有(CD8,CD28)跨膜結構域和(CD28,4-1BB)信號傳導結構域的CAR的產生
測試的各個CAR已經顯示相似的擴增、細胞毒性和Th1細胞毒性。CD19-BB-z已經顯示了較低的Th2細胞因子產生;在體內,它在控制小鼠中的疾病方面是有效的(Molecular Therapy 17(8):1453–1464,2009)。不過,由于細胞在沒有抗原性刺激的情況下在體外存活的事實而存在憂慮。
下文在表2中顯示了臨床中的CAR。在一些實施方案中,本發明的CAR沒有如下文表2中顯示的特定構建體。或者,在一些實施方案中,可以使用本發明的方法產生具有下文表2中提到的CAR特征,不過與目前在臨床中使用的CAR截然不同的CAR的另一種變化。
表2:臨床中的CAR
圖3中說明了具體的CAR構建體設計。如圖3中顯示,產生使用CD19scfv、CD8a鉸鏈或IgG4Fc莖部、CD8跨膜(TM)或CD28TM或CD137TM和經CD28或CD137膜內結構域連同CD3ζ膜內結構域信號傳導的組合的各個CAR的示意圖。
然后,將圖3中顯示的CAR構建體克隆到含有SIM和FRA標簽的睡美人質粒中以允許在使用常見的CVseq7引物擴增時在競爭再增殖研究中追蹤。圖4中顯示了睡美人追蹤質粒。
使用Amaxa Nucleofector II將圖3中顯示的CAR構建體電穿孔到T細胞中,并且在存在細胞因子(IL2,IL-21)的情況下與aAPC共培養28天。顯示了電穿孔后1天(第1天)和與aAPC共培養28天(第28天)后的CAR表達。顯示了CD3和CAR的點圖,其中使用CD3和抗CD19scfv特異性Ab區分T細胞和CAR。圖5中顯示了CAR表達結果。
隨時間對圖3中顯示的CAR構建體評估CAR表達持續28天,并且顯示。在21天后,大多數培養物具有>80%CAR表達。圖6中顯示了CAR表達動力學。
在與aAPC共培養28天后顯示了用來自圖3的CAR核轉染的培養物中的CD4和CD8T細胞的表達百分比。圖7中顯示了這些表型結果。
在共培養28天后,對CAR+T細胞(表達圖3中描述的CAR)評估屬于記憶(CD45RA、CCR7、CD27)、活化(CD69、HLA-DR)、細胞毒性(穿孔蛋白、粒酶B)、耗竭/衰老(CD57、KLRG1、PD1)、和粘附(CD39、CD150)的標志物表達。圖8A-B中顯示了此延伸表型的結果。
使用蛋白質印跡對CAR+T細胞(表達圖3中描述的CAR)評估CD3ζ的表達。在變性條件下運行細胞裂解物,轉移,并且使用一級小鼠抗人CD3ζmAb和偶聯有HRP的山羊抗小鼠IgG使用SuperSignal West Femto Maximum Sensitivity底物測量嵌合CD3ζ的表達。相對于CAR構建體的大小,觀察到52、71和78kD的嵌合CD3ζ條帶。圖9中顯示了這些蛋白質印跡結果。
在第1天和此后每7天持續28天,用K562aAPC刺激用CAR構建體(圖3中描述)電穿孔的T細胞。在每個刺激循環結束時,使用臺盼藍排除方法計數細胞,并且在CD3和CAR表達方面進行表型分析。圖10中顯示的圖描繪了隨時間總共、CD3、和CAR+T細胞的推斷的細胞計數。
測量細胞的擴增。通過與(電穿孔后)第1天比較計數在共培養的第14天、第21天和第28天計算總細胞(圖11)和CAR+(圖12)T細胞的倍數擴增。圖11和圖12中顯示了結果。
對CAR表達T細胞(CAR+T細胞)測量細胞毒性。在標準的4小時鉻釋放測定法中,與CD19-EL-4相比,對CAR+T細胞(表達圖3中描述的CAR)評估其針對CD19+腫瘤靶標(Daudiβ2m、NALM-6和CD19+EL-4)的細胞毒性。在圖13、圖14、圖15和圖16中顯示了結果。
胞內IFN-γ產生。在存在蛋白質轉運抑制劑的情況下將CAR+T細胞(表達圖3中描述的構建體)與(CD19+和CD19-)刺激細胞一起溫育4-6小時,固定,透化,并且用IFN-γ特異性mAb染色。PMA-離子霉素用作陽性對照。圖17、圖18、圖19和圖20中顯示了胞內IFN-γ產生的結果。
針對SB11轉座酶的PCR。在熱循環儀中使用SB11特異性引物擴增從CAR+T細胞分離的DNA(圖3)。GAPDH用作持家基因,并且線性化pCMV-SB11質粒,來自表達SB11的Jurkat細胞的基因組DNA用作陽性對照。CAR-細胞(無DNA)用作陰性對照。圖21中顯示了這些PCR結果。
使用定量PCR(qPCR)測量CAR拷貝數。通過使用對IgG4Fc莖部和反向/同向重復序列(IR/DR)特異性的引物和探針擴增基因組DNA來評估細胞中的CAR轉基因(圖3中顯示的CAR構建體)的整合數目。RNA酶P基因用作內部對照,并且表達單拷貝CAR的Jurkat細胞系用于產生標準曲線。圖22中顯示了結果。
接著,測量CAR+T細胞的自主生長的存在或缺乏。通過在缺乏細胞因子和aAPC的情況下培養T細胞來監測并測量CAR+T細胞(表達圖3中顯示的CAR構建體)的異常生長。每7天計數細胞,并且計算并繪圖活細胞/死亡細胞百分比(從第1天起)。如圖23中顯示,觀察到超過80%的T細胞到第14天是死亡的,顯示缺乏自主生長。
各個CAR可以進行表達(>80%),擴增(約1010),并且在相似程度上有細胞毒性(約60%,Daudi)。支架結構域(IgG4或CD8α)用于建立CAR,并且不影響表達或效力。跨膜結構域(CD8,CD28)不影響效力。4-1BB跨膜結構域(216)影響表達(抗scFv Ab),但是不影響細胞毒性和細胞因子產生。信號傳導結構域、CD28和4-1BB的組合沒有疊加效應。CAR+T細胞展現出記憶/效應子表型。與其它CAR相比,僅含有4-1BB結構域(212、214、217)的CAR具有更高的CCR7表達。細胞表達記憶(CD27hi、CD45RAhi、CCR7lo)、活化(CD69med、HLA-DRhi)、細胞溶解(粒酶hi,穿孔蛋白lo),和粘附(CD39hi,CD150lo)的標志物,但是觀察到可忽略不計量的抑制性標志物(CD57、PD1、KLRG1)。所有CAR,包括含有4-1BB結構域域的CAR,缺乏SB11轉座酶,并且不自身增殖。
實施例4
含有CD3-ζ的CAR的產生
在本實施例中提供了含有CD3ζ的CAR。圖24中顯示了CAR設計的總圖。如圖24中顯示,顯示了CAR設計(圖24,右)與抗體分子(圖24,左)的比較。
圖25中顯示了CD3ζ序列。圖25中顯示了CD3ζ及其同等型的序列。CAR設計包括CD3ζ(同等型1),其形成膜內結構域信號傳導部分之一并且具有三個ITAM。
圖26和圖27中顯示了具體的CAR構建體。圖26顯示了具有長(IgG4)、中等(CD8a鉸鏈)和小(IgG 12aa)莖部,經由CD28或CD137膜內結構域進行信號傳導的CD19特異性CAR的示意圖。圖27中顯示了具有不同莖部和信號傳導的CAR分子的命名。
測量CAR表達。在電穿孔后1天(第1天)和在aAPC上共培養28天(第28天)后測量CAR(如圖26中描述)的表達。圖28中顯示了CD3和CAR(如通過CD19scfv特異性mAb測量)的點圖。
對CAR測量擴增動力學。在7天刺激循環中在aAPC上共培養用CAR構建體(圖26中顯示)電穿孔的T細胞。對細胞計數,并且評估CD3和CAR的表達。圖29和圖30中顯示了結果。
測量CAR+T細胞的細胞毒性。在28天共培養結束時,在鉻釋放測定法中評估CAR+T細胞(表達圖26中顯示的構建體)的針對腫瘤靶標的細胞毒性。如圖31中顯示,對針對CD19+和CD19-腫瘤靶標的CD19RCD28(CAR 194)和CD19RCD137(CAR 217)CAR以各個效應物與靶標比率測量細胞毒性百分比。如圖32中顯示,獲得在20:1的E:T比率時CAR+T細胞(表達圖26中顯示的CAR構建體)對CD19+EL-4的裂解百分比的數據。測量CAR+T細胞(表達圖26中顯示的構建體)上的CD27、CD62L、CD28和CCR7的表達百分比,并且在圖33中顯示了結果。
對CAR+T細胞測量胞內細胞因子產生。在存在蛋白質轉運抑制劑的情況下將刺激細胞(CD19+和CD19-)與CAR+T細胞(表達圖26中顯示的CAR)一起溫育4小時,并且用IFN-γ和IL-2mAb染色。PMA-離子霉素充當陽性對照,并且單獨的T細胞充當陰性對照。圖34顯示了刺激后的IFN-γ生成細胞的百分比。圖35顯示了與細胞刺激混合物(PMA-離子霉素)一起溫育后IFN-γ和或IL-2生成細胞的分解。
對CAR+T細胞測量自主生長的存在或缺乏。對CAR+T細胞(表達圖26中描述的CAR)評估其在缺乏外部刺激(細胞因子和aAPC)的情況下缺乏異常生長持續18天。在18天結束時,超過80%的細胞是死亡的,顯示了缺乏不想要的生長。如圖36中顯示,觀察到自主生長的缺乏。
在CAR+T細胞中測量CAR拷貝數。通過qPCR使用對IgG4-Fc和IR/DR區特異性的引物/探針評估整合的CAR分子的拷貝數。如圖37中顯示,使用IR/DR探針觀察到整合的CAR拷貝數(圖26中顯示的CAR)。如圖38和圖39中顯示,在CAR構建體(圖3中顯示的兩種CAR構建體和圖26中顯示的CAR構建體)的表和圖形形式中提供了CAR拷貝數數據的匯編,如在兩個不同實驗中測試(P491;C714和GCR357861)。
這些數據顯示了可以在如本文中描述的培養系統中體外表達并培養具有各種間隔區的CAR。觀察到所有CAR具有相似的CAR表達。在具有CD8鉸鏈區的CAR中觀察到CD19+EL-4s的最大細胞毒性。對測試的所有CAR觀察到相似的CD62L和CD28表達。除了含有IgG4-Fc莖部的CAR外,在所有CAR中觀察到如通過CAR拷貝數測量的高整合頻率。通過PCR觀察到自主生長和SB11的缺乏。與先前的報告形成對比,在CAR中包含12aa間隔區在這些研究中沒有賦予改善的功能性。
實施例5
從主要組分快速裝配CAR
與臨床級CD19RCD28mζCAR+T細胞(CG CAR)平行,發明人使用EZ CAR平臺產生經由嵌合CD28/CD3-ζ活化的CD19特異性CAR。將臨床級CD28/CD3-ζ和EZ CAR CD19RCD28mζCAR序列兩者插入睡美人轉座子載體中,并且電穿孔到T細胞中。在電穿孔后,在存在CD19+人工抗原呈遞細胞(又稱作活化和增殖細胞,或AaPC)的情況下培養T細胞,用于T細胞的抗原特異性擴增。通過流式細胞術(Fc+表達)每周測量T細胞表面中的CAR表達,顯示了臨床級CD19CAR T細胞和EZ CD19CAR T細胞中的相似CAR表達。還實施鉻釋放測定(CRA)以評估通過EZ CAR平臺產生的T細胞CD19CAR+針對腫瘤細胞的殺傷功能。在溫育4小時后,通過EZ CAR T細胞觀察到特異性細胞裂解的百分比為52%,并且通過CG CAR T細胞為49%。
這些結果證明了使用這些方法產生功能性CAR+T細胞。然后,發明人使用與含有CAR分子的以下三種組分的質粒文庫組合的如上文描述的方法進行CAR的快速產生:(i)抗CD19scFv,(ii)具有不同尺寸的5種鉸鏈(長-IgG4a和IgG4ΔEQ,中等-CD8α,短-t-20AA和t-12AA)和(iii)具有CD3ζ結構域的7種信號傳導結構域(CD27、CD28、CD28ΔY173→F173、CD134、CD137、CD278)的不同組合。用含有CAR轉基因的質粒轉染HEK 293細胞用于篩選27種不同CAR構建體以確保細胞表面中的CAR蛋白的表達。使用iQueTM篩選器(Intellicyt,Albuquerque,NM)(一種高通量流式細胞儀)進行個別CAR分子的高通量測試,其中使用表達熒光粒酶B報告物或GFP的工程化靶細胞進行細胞毒性測定。圖40A-E中顯示了結果。
使用IntelliCyt’s iQueTM,進行額外的實驗以篩選CAR分子。iQueTM使用高通量流式細胞術,一種通過使用復用(multiplexing)性能和逐個細胞分析(cell-by-cell analysis)研究大群體產生信息的補充技術。發明人采用此技術來告知用CAR組修飾的T細胞的治療潛力。可以針對活力,及活化信號(例如,CD25的上調)、細胞因子釋放、和殺傷方面對來自孔的T細胞染色。因此,發明人調適iQue篩選器,并且利用其進行復用的基于珠粒的細胞因子檢測和基于細胞的測定的能力。獲得的結果指示此技術可以用于測試通過EZ CAR平臺產生的大量不同CAR T細胞。使用IntelliCyt’s iQueTM產生數據,其中對CAR T細胞的2個群體評估其殺傷靶細胞的能力。圖41A-B和圖42中顯示了結果。這些結果證明了CAR分子有活性,并且iQueTM方法可以有效用于評估CAR活性。
***
本文中公開和要求保護的所有方法可以根據本公開內容進行和執行而無需過度實驗。雖然本發明的組合物和方法已經按照優選的實施方案描述,但是對于本領域技術人員將明顯的是,可以在不偏離本發明的構思、精神和范圍的前提下對方法和在本文中描述的方法的步驟中或步驟的順序中應用變化。更具體地,將明顯的是,可以用化學和生理學相關的某些藥劑替代本文中的描述的藥劑,同時將實現相同或相似的結果。認為對于本領域技術人員明顯的所有此類類似的替代和修飾在本發明的精神、范圍和構思內,如所附權利要求書限定。
參考文獻
通過引用方式將以下參考文獻明確并入本文,其程度為它們提供對本文中列出的那些補充性的示例性程序或其它詳情。
美國公開No.2009/0017000
美國公開No.2009/0004142
美國專利No.6,225,042
美國專利No.6,355,479
美國專利No.6,362,001
美國專利No.6,410,319
美國專利No.6,790,662
美國專利No.7,109,304
WO2007/103009
Ahmed and Cheung,FEBS Lett.2014 Jan 21;588(2):288-97.
Altenschmidt et al.,Adoptive transfer of in vitro-targeted,activated T lymphocytes res ults in total tumor regression,J Immunol.1997 Dec 1;159(11):5509-15.
Audet et al.,Sci Rep.2014 Nov 6;4:6881.
Berry et al.Adoptive immunotherapy for cancer:the next generation of gene-engineered immune cells.Tissue Antigens.2009 Oct;74(4):277-89.
Brocker et al.,Adv.Immunol.,68:257,1998.
Curtin et al.,MAbs.2015 Jan 2;7(1):265-75.
et al.,Drug Metab Dispos.2015 Jan;43(1):42-52.
Davies JK,Singh H,Huls H,Yuk D.Lee DA,et al.(2010)Combining CD19 redirection and alloanergization to generate tumor-specific human T cells for allogeneic cell therapy of B-cell malignancies.Cancer Res 70:3915-3924.
de Weers et al.,J Immunol.2011 Feb 1;186(3):1840-8.
Duong et al.,(2013)Engineering T Cell Function Using Chimeric Antigen Receptors Identified Using a DNA Library Approach.PLOS ONE 8(5):e63037.
Eshhar et al.,Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors.Proc Natl Acad Sci U S A.;90(2):720-4,1993.
Eshhar,Tumor-specific T-bodies:towards clinical application.Cancer Immunol Immunother.1997 Nov-Dec;45(3-4):131-6.1997
Fitzer-Attas et al.,Harnessing Syk family tyrosine kinases as signaling domains for chimeric single chain of the variable domain receptors:optimal design for T cell activation.J Immunol.1998 Jan1;160(1):145-54.1998
Funakoshi et al.,Cancer Treat Rev.2014 Dec;40(10):1221-9.
Gerber et al.,Clin Cancer Res.2011 Nov 1;17(21):6888-96.
Goldberg et al.,J Clin Oncol.2014 May 10;32(14):1445-52.
Gross et al.,Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity.Proc.Natl.Acad.Sci.USA,86:10024-10028,1989.
Gross et al.(1992)Endowing T cells with antibody specificity using chimeric T cell receptors.FASEB J.1992 Dec;6(15):3370-8.
Hackett et al.,A transposon and transposase system for human application,Mol Ther.2010 Apr;18(4):674-83).
Hekele et al.Growth retardation of tumors by adoptive transfer of cytotoxic T lymphocytes reprogrammed by CD44v6-specific scFv:zeta-chimera.Int J Cancer.1996 Oct 9;68(2):232-8,1996.
Huls et al.,Clinical Application of Sleeping Beauty and Artificial Antigen Presenting Cells to Genetically Modify T Cells from Peripheral and Umbilical Cord Blood.J.Vis.Exp.,doi:10.3791/50070,2013.
Huls et al.“Clinical application of Sleeping Beauty and artificial antigen presenting cells to genetically modify T cells from peripheral and umbilical cord blood”J Vis Exp.2013 Feb 1;(72):e50070.
Humbler-Baron and Baron,Immunol Cell Biol.2015 Feb 10.doi:10.1038/icb.2014.120.
Hwu et al.(1995)In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell rcccptor gcnes.Cancer Res.1995 Aug 1;55(15):3369-73.
Ikeda et al.,Clin Cancer Res.2009 Jun 15;15(12):4028-37.
Jabbour et al.,Am J Hematol.2014 Nov 18.
Kaufmann et al.,Hum Pathol.1997 Dec;28(12):1373-8.
Kaufman et al.,Br J Haematol.2013 Nov;163(4):478-86.
Kim DW,Uetsuki T,Kaziro Y,Yamaguchi N,Sugano S(1990)Use of the human elongation factor l alpha promoter as a versatile and efficient expression system.Gene 91:217-223.
Kim et al.,2004,Nature,Vol.22(4),pp.403-410.
Kim et al.,Immunology.2010 Aug;130(4):545-55.
Kong et al.,Leuk Res.2014 Nov;38(11):1320-6.
Krebs et al.,T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice.Gastroenterology.2013 Aug;145(2):456-65.
Le Garff-Tavernier et al.,Haematologica.2014 Dec 31.
Lennard S.,Standard protocols for the construction of scFv libraries.Methods Mol Biol.2002;178:59-71.
Leung,Molecular Imaging and Contrast Agent Database(MICAD),Bethesda(MD):National Center for Biotechnology Information(US):2004-2013.2010 Mar 25
Leung 2011,IRDye800CW-anti-CD105 TRC105 chimeric monoclonal antibody.Molecular Imaging and Contrast Agent Database(MICAD).Bcthesda(MD):National Center for Biotechnology Information(US);2004-2013.2011 Dec 01
Maiti et al.,Sleeping beauty system to redirect T-cell specificity for human applications.J Immunother.36(2):112-23,2013.
Manero et al.,Haematologica.2013 Feb;98(2):217-21.
Molecular Therapy 17(8):1453-1464,2009.
Moritz et al.(1994)Cytotoxic T lymphocytes with a grafted recognition specificity for ERBB2-expressing tumor cells.Proc Natl Acad Sci U S A.1994 May 10;91(10):4318-22.
Patel et al.,Anticancer Res.2008 Sep-Oct;28(5A):2679-86.
Qiu et al.,PLoS Negl Trop Dis.2012;6(3):e1575.
Remington′s Pharmaceutical Sciences,16th Ed.,Mack,ed.(1980)
Rushworth et al.,(2014)“Universal Artificial Antigen Presenting Cells to Selectively Propagate T Cells Expressing Chimeric Antigen Receptor Independent of Specificity”J Immunother.May;37(4):204-13.
Sarup et al.,Mol Cancer Ther.2008 Oct;7(10):3223-36.
Schneider,J.Embryol.Exp.Morph.1972 Vol 27,pp.353-365
Schultz-Thater et al.,Br J Cancer.2000 Jul;83(2):204-8.
Shin et al,Immune Netw.2011 Apr;11(2):114-22.
Singh et al.,Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system.Cancer Res.,68:2961-2971,2008.
Singh H,Figliola MJ,Dawson MJ,Huls H,Olivares S,et al.(2011)Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies.Cancer Res 71:3516-3527.
Singh et al.“A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19.”Immunol Rev.2014 Jan;257(1):181-90.
Singh et al.,“Manufacture of T cells using the Sleeping Beauty system to enforce expression of a CD 19-specific chimeric antigen receptor.”Cancer Gene Ther.2015 Jan 16.
Stancovski et al.(1993)
Stynen et al.,Fungal Cell Wall and Immune Response,NATO ASI Series Volume 53,1991,pp 181-193.
Sun et al.,Cell Mol Immunol.2007 Jun;4(3):209-14.
Turtle et al.,“Artificial antigen-presenting cells for use in adoptive immunotherapy”Cancer J.2010 Jul-Aug;16(4):374-81.
Vcrcl,Iht J Cancer.2002 May 20;99(3):396-402.
Vincent and Samuel,Journal of Immunological Methods,Volume 165,Issue 2,15 October 1993,Pages 177-182.
Wang et al.,Immunology.2015 Feb;144(2):254-62.
Weijtens et al.(1996)Single chain Ig/gamma gene-redirected human T lymphocytes produce cytokines,specifically lyse tumor cells,and recycle lyric capacity.J Immunol.1996 Jul 15;157(2):836-43.
Wieczorek et al.,Genetically Modified T Cells for the Treatment of Malignant Disease.Transfus Med Hemother.2013 Dec;40(6):388-402.
Winiarska et al.,MAbs.2014;6(5):1300-13.
Zhuang et al.,Cancer Cell Int.2014 Nov 30;14(1):109.
Zhang et al.,Angiogenesis.2002;5(1-2):35-44.
- 還沒有人留言評論。精彩留言會獲得點贊!
- 金黃色葡萄球菌腸毒素C2的核酸適配體C203及其篩選方法和應用與制造工藝
- 一種全人源化EGFRvIII嵌合抗原受體T細胞及其制備方法與制造工藝
- 嵌合抗原受體及制備方法與制造工藝
- 檢測外周血CAR?T細胞的TaqMan實時熒光定量PCR試劑盒的制造方法與工藝
- CD20特異性嵌合抗原受體及其應用的制造方法與工藝
- 一種用于辣根過氧化物酶染色的染色劑及其制備方法、染色組合、應用及標記酶染色試劑盒與制造工藝
- 對SSEA4抗原具有特異性的嵌合抗原受體的制造方法與工藝
- 具有針對CD79和CD22的特異性的分子的制造方法與工藝
- 具有針對CD45和CD79的特異性的分子的制造方法與工藝
- 一種檢測嵌合抗原受體T細胞對肝癌細胞抑制作用的模型和方法與制造工藝