藥物遞送增強劑的制作方法
上皮細胞構成了藥物遞送的屏障。例如,口服給藥的治療藥物的吸收可能受到腸上皮的限制,并且通過吸入而遞送的治療藥物的吸收可能受到肺上皮的限制。同樣,遞送至其他上皮部位(如直腸腔、口腔、陰道腔、鼻腔)的、或者通過局部施用于皮膚的治療藥物的吸收可類似地受到上皮屏障的限制。該屏障對包括小分子、肽、蛋白質、疫苗和核酸在內的大范圍的藥物的遞送提出了挑戰。已經進行了各種嘗試來減輕由上皮細胞提供的屏障對藥物吸收的影響。然而,許多這些嘗試涉及使用有意或作為非預期的副作用損傷上皮的試劑。這種損傷可能導致材料不受控制地通過上皮,并且可能與由細胞損傷引起的免疫學危險信號相結合,可能導致上皮的不期望的炎癥。因此,需要幫助治療劑通過上皮表面(優選以可控的、暫時的且不引起無法接受的細胞損傷的方式)的改進的技術(Brayden&Maher(2000)Therapeutic Delivery 1(1):5-9)。
由上皮提出的對于藥物吸收的挑戰已經導致了某些藥物需要通過除了穿過上皮以外的途徑來遞送,例如,通過皮下注射。這帶來了許多缺點,包括降低的患者可接受性,更低的患者的依從性,以及與使用和貯存液體配方、提供注射裝置和以安全和負責的方式處置用過的注射裝置相關的更高成本。
上皮提供了對藥物吸收的屏障,因為其包括通過緊密連接而彼此連接的細胞層,所述緊密連接提供了對許多物質的密閉,但是其能夠瞬時開啟。存在有能夠更好地控制緊密連接的開啟以幫助治療藥物遞送的需要。對于開啟緊密連接的問題已經提出了多種解決方案。這些努力已經引起了兩組分子的鑒定:非天然的、延長的氨基酸(NEAA)和短鏈脂肪酸(SCFA)。NEAA已由Emisphere公司最初開發,該公司已證明了藥物攝取中的邊際改善,但是尚未提交新藥的監管批準申請。一種SCFA,癸酸鈉用在經批準的直腸栓劑產品中以改善氨芐青霉素的攝取。然而,懷疑改善攝取的機制是通過粘膜上皮的局部損傷(Maher等人,(2009)Adv.Drug Deliv.Rev.61:1427-1449)。
特別感興趣的是口服遞送胰島素、胰高血糖素樣肽1(GLP-1)和其它相關分子用于治療糖尿病患者、肥胖癥、抑制食欲和改善碳水化合物代謝。這樣的化合物特別需要口服遞送途徑,因為它們通常被施用數月、數年或更長時間,這意味著避免注射可顯著提升患者的可接受性并潛在地降低成本,并且還因為在自然中這些腸激素通過肝門靜脈進入受試者的血流,所以穿過腸屏障遞送將更接近地模擬生理遞送途徑。
本發明基于發現了對于上皮的緊密連接的受控開啟有用的化合物。
技術實現要素:
根據發明的第一方面,提供一種肽,其包含根據式1的序列:
X4-X5-X6-X7-X8
式1(SEQ ID NO:1)
其中:
X4為帶負電的氨基酸殘基,例如pTyr、Asp或Glu;
X5為小的疏水性氨基酸殘基,例如Val或Ala;
X6為帶正電的氨基酸殘基,例如Lys或Arg;
X7為親水性氨基酸殘基,例如Tyr、Phe、Thr或Ser;并且
X8為帶負電的氨基酸殘基,例如pTyr、Asp或Glu;
或者所述肽包含根據式2的序列:
X5-X6-X7-X8
式2(SEQ ID NO:2)
其中:
X5為任意氨基酸殘基;
X6為Tyr;
X7為Gln;并且
X8為Tyr;
或者所述肽包含根據式1或式2的序列的逆-反(retro-inverso)形式,其中所有氨基酸殘基均為D-構型并且肽序列順序反轉。
根據發明的第二方面,提供一種藥物組合物,其包含一種肽以及藥學上可接受的載體,所述肽包含根據式1的序列:
X4-X5-X6-X7-X8
式1(SEQ ID NO:1)
其中:
X4為帶負電的氨基酸殘基,例如pTyr、Asp或Glu;
X5為小的疏水性氨基酸殘基,例如Val或Ala;
X6為帶正電的氨基酸殘基,例如Lys或Arg;
X7為親水性氨基酸殘基,例如Tyr、Phe、Thr或Ser;并且
X8為帶負電的氨基酸殘基,例如pTyr、Asp或Glu;
和/或所述肽包含根據式2的序列:
X5-X6-X7-X8
式2(SEQ ID NO:2)
其中:
X5為任意氨基酸殘基;
X6為Tyr;
X7為Gln;并且
X8為Tyr;
或者其中根據式1和/或式2的序列為逆-反構型,其中所有氨基酸殘基均為D-構型并且肽序列順序反轉。
根據發明的第三方面,提供根據發明第一方面的肽或者根據發明第二方面的藥物組合物,其用作藥物。
根據發明的第四方面,提供根據發明第一方面的肽或者根據發明第二方面的藥物組合物在制備藥物中的用途。
根據發明的第五方面,提供一種打開上皮表面的緊密連接的方法,其包括將有效量的根據發明第一方面的肽或者根據發明第二方面的藥物組合物施用于上皮。
根據發明的第六方面,提供一種遞送藥劑穿過上皮表面的方法,其包括將所述藥劑與根據發明第一方面的肽聯合施用,或者作為根據發明第二方面的藥物組合物的一部分進行施用。
附圖說明
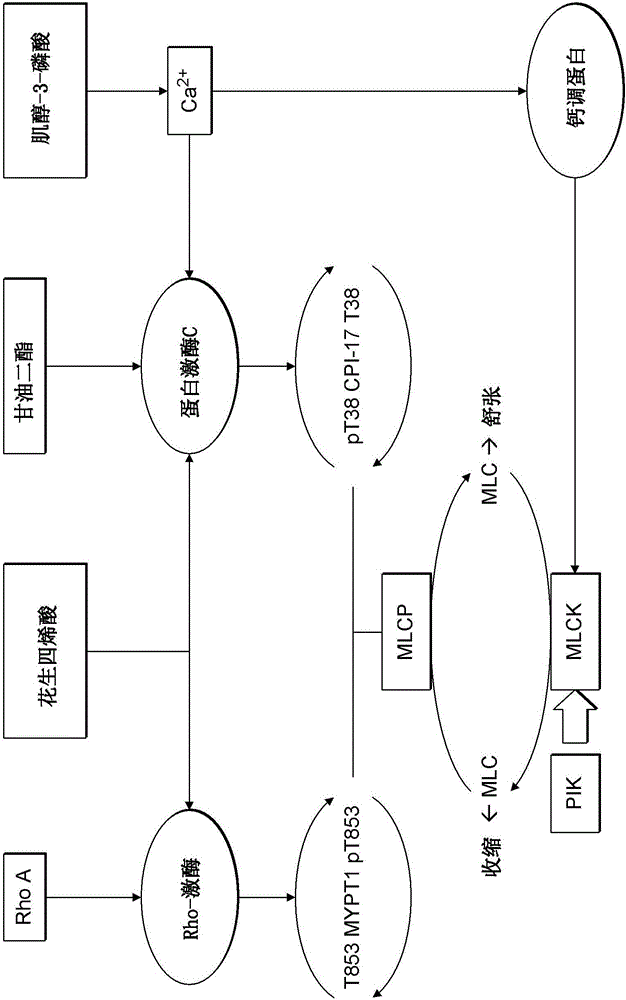
圖1:已知調節肌球蛋白輕鏈(MLC)磷酸化狀態的磷酸酶和激酶的組織。能夠使MLC磷酸化的肌球蛋白輕鏈激酶(MLCK),通過細胞質鈣(Ca2+)-鈣調蛋白水平的增加而被活化。MLCK的活化形式能夠被小的、膜可透過的肽激酶抑制劑(PIK)抑制。肌球蛋白輕鏈磷酸酶(MLCP)復合物的活性受到被稱為CPI-17和MYPT1的兩種調節亞基的控制。這些調節亞基的作用依次分別受到蛋白激酶C和Rho激酶活性的控制。
圖2:緊密連接(TJ)開啟/關閉和MLC磷酸化/去磷酸化的動態調節。在天然狀態中,MLCP是活化的,這使MLC保持在去磷酸化的狀態并且TJ結構在關閉狀態,其中存在最少的細胞旁(相鄰上皮細胞之間)溶質通量(flux)。MLCK的持續活化或MLCP的抑制將導致MLC磷酸化狀態增加和細胞旁溶質通量的增加。
圖3:MYPT1的殘基1-299與PP1在一個表面相互作用,而CPI-17與另一個面相互作用。晶體結構的帶狀描述說明了MYPT1與CPI-17預計如何在平滑肌細胞中相互作用。這些相互作用可能不同于那些分化的上皮細胞。
圖4:[A]RRVEVKYDRR(肽640,SEQ ID NO:3)相對于沒有肽的對照以1mM和5mM對Caco-2細胞單層TEER(跨上皮電阻)的作用的時間過程。[B]至多5mM沒有肽細胞毒性——MMT試驗、半胱天冬酶的活化等等。
圖5:RRVEVKYDRR(肽640,SEQ ID NO:3)和RKAKYQYRRK(肽250,SEQ ID NO:4)相對于對照肽(均以5mM加入)對TEER[左]和4kDa的熒光素標記的葡聚糖(4FD)轉運[右]的作用。在0、15、30、45、60、75、90和120分鐘進行測量。在45分鐘時,所有頂部的培養基用4FD但沒有肽的培養基替換。在70分鐘時,所有頂部的培養基用4FD加上在沖去之前存在的相同肽的培養基來重新裝滿。每個時間點的值表示兩個Caco-2單層的平均值;未進行統計學分析。
圖6:在應用肽250或肽640之后穿過Caco-2單層的(A)絕對TEER和(B)初始TEER%。箭頭表示當沖去肽并用PBS代替時的點。對照為沒有肽的PBS。對于對照,n=6,并且,對于所有肽組,n=5。數據顯示為平均值±SD。
圖7:在應用葡聚糖與肽250或肽640之后,在基底外側室中發現的(A)4kDa的葡聚糖的累積量和(B)70kDa的葡聚糖的累積量。對于對照,n=6,并且,對于所有肽組,n=5。數據顯示為平均值±SD。
圖8:(A)在雄性Wistar大鼠中S.C.注射胰島素之后的初始血糖%。(B)在腸內注射肽250與30IU/kg的胰島素之后的初始血糖水平%和(C)在腸內注射肽640與30IU/kg胰島素之后的初始血糖水平%。對于所有數據組,n=3。數據顯示為平均值±SD。
圖9:腸腔內注射PIP肽瞬時增加了pMLC含量并在體內在非糖尿病大鼠腸組織中增強了胰島素的細胞旁攝取。(A)代表性半定量蛋白質印記分析,其用于評估相對于總的MLC含量,肌球蛋白輕鏈(MLC)在Ser19處磷酸化(pMLC-Ser19)的程度的變化。(B)ILI注射PIP肽增加了pMLC的含量。以設定的濃度在頂部應用PIP肽之后,在選定的時間點大鼠腸組織中的MLC與pMLC-Ser19水平之比。*p<0.05。(C)在ILI注射之后,通過PIP肽增強的發熒光的(Cy3-標記的)胰島素的細胞旁轉運。細胞核是DAPI染色的(藍色)。
圖10:PIP肽250和640以不同動力學增強了非糖尿病大鼠的腸腔攝取人胰島素。(A)在ILI注射PIP肽與30IU/kg的胰島素之后,從門靜脈采集的血液的血清樣本中胰島素的時間-濃度曲線。單因素方差分析(one-way ANOVA)顯示數據組彼此顯著不同(p<0.01)。Bonferroni后測試顯示,胰島素與肽250(p<0.01)和胰島素與肽640(p<0.05)與僅有胰島素顯著不同。(B)在ILI注射PIP肽與30IU/kg的胰島素之后,從尾靜脈采集的血清樣本中胰島素的時間-濃度曲線。當使用單因素方差分析進行比較時在組之間沒有顯著的差別。(C)在SC注射3IU/kg的人胰島素之后,從非糖尿病大鼠的尾部和門靜脈采集的血液的血清樣本中胰島素的時間-濃度曲線。對每個治療組來說n=3,數據為平均值±SD。
圖11:PIP肽250和640以不同動力學增強了非糖尿病大鼠的腸腔攝取人胰島素。(A)在ILI注射PIP肽與30IU/kg的胰島素之后,從門靜脈采集的血液的血清樣本中胰島素的時間-濃度曲線。單因素方差分析(one-way ANOVA)顯示數據組彼此顯著不同(p<0.01)。Bonferroni后測試顯示,胰島素與肽250(p<0.01)和胰島素與肽640(p<0.05)與僅有胰島素顯著不同。(B)在ILI注射PIP肽與30IU/kg的胰島素之后,從尾靜脈采集的血清樣本中胰島素的時間-濃度曲線。當使用單因素方差分析進行比較時在組之間沒有顯著的差別。(C)在SC注射3IU/kg的人胰島素之后,從非糖尿病大鼠的尾部和門靜脈采集的血液的血清樣本中胰島素的時間-濃度曲線。對每個治療組來說n=3,數據為平均值±SD。
定義
多肽:一種單體為氨基酸殘基通過酰胺鍵連接在一起的聚合物。當氨基酸為α-氨基酸時,能夠使用L-光學異構體或者D-光學異構體。本發明涉及L-型異構體和D-型異構體二者。特別地,但非排它地,當肽包含與式1或2中所指定的序列相反的序列時,發明優選地涉及D-型異構體。這樣的肽是指為“逆-反”形式。本文所用的術語“多肽”或“蛋白質”包括任何氨基酸序列,并且包括修飾的序列如糖蛋白以及使用天然存在和非天然存在的氨基酸的序列。術語“多肽”涵蓋了天然存在的蛋白質,以及重組或合成制備的蛋白質,多肽的鹽、溶劑合物和衍生物。本發明的肽優選在其C末端酰胺化。
發明的肽的鹽或溶劑合物優選為其中補償離子(counterion)或相關溶劑是藥學上可接受的那些。然而,具有非藥學上可接受的補償離子或相關溶劑的鹽和溶劑合物也在本發明的范圍之內,例如,用作中間體。
肽活性
與發明所有方面相關的肽優選為在其增強細胞旁轉運的能力方面具有活性。這可根據本文詳述的方法之一(特別是在實施例中)來測量。
鹽和溶劑合物
根據發明合適的鹽包括那些與有機的或無機的酸或堿形成的。藥學上可接受的酸加成鹽包括那些與鹽酸、氫溴酸、硫酸、硝酸、檸檬酸、酒石酸、乙酸、磷酸、乳酸、丙酮酸、乙酸、三氟乙酸、琥珀酸、高氯酸、富馬酸、馬來酸、乙醇酸、乳酸、水楊酸、草酰乙酸、甲磺酸、乙磺酸、對甲苯磺酸、甲酸、苯甲酸、丙二酸、萘-2-磺酸、苯磺酸和羥基乙磺酸形成的。其他酸如草酸,雖然其本身不是藥學上可接受的,但在獲得發明的化合物及其藥學上可接受的鹽時可用作中間體。與堿形成的藥學上可接受的鹽包括銨鹽、堿金屬鹽(例如鉀鹽和鈉鹽)、堿土金屬鹽(例如鈣鹽和鎂鹽)、以及與有機堿形成的鹽(例如二環己基胺和N-甲基-D-葡糖胺(glucomine))。
有機化學領域的技術人員將理解到,許多有機化合物能夠與它們在其中發生反應或它們從中沉淀或結晶的溶劑形成復合物。這樣的復合物被稱為“溶劑合物”。例如,與水的復合物被稱為“水合物”。本發明提供發明化合物的溶劑合物。
肽合成
發明的肽可通過任何對制備肽來說合適的技術來制備,包括但不限于常規的方法學,例如,從單獨的氨基酸來合成,特別是用全自動肽合成儀的逐步合成;天然肽的修飾;或者重組制造技術。
雖然可能將發明的肽單獨施用,但是優選其存在于藥物組合物中。因此,發明提供一種藥物組合物,其包括發明的肽以及藥學上可接受的載體。發明的藥物組合物可采取如下所述的藥物配方的形式。
藥物配方
與發明有關的藥物配方包括那些適用于口服、腸外吸入(parenteral inhalation)(包括可通過各種類型的計量劑量、加壓氣溶膠、霧化器或吹入器的手段產生的細顆粒粉劑或霧劑)、直腸和局部(包括皮膚、透皮、粘膜、口腔、舌下或眼內)給藥,盡管最合適的途徑可能取決于例如受體的狀況和病癥。
配方可方便地以單位劑量的形式存在,并且可通過任何藥學領域熟知的方法制備。所有方法都包括使活性成分與構成一種或多種助劑的藥物載體相結合的步驟。通常,配方通過使活性成分與液體載體或細碎的固體載體或兩者均勻且緊密地結合來制備,然后,如有必要,使產品成形為所需的配方。
適用于口服給藥的本發明配方可作為分別包含預定量的活性成分的離散單元如膠囊、扁囊劑或片劑而存在;作為粉末或顆粒而存在;作為含水液體或非含水液體中的溶液或懸浮液而存在;或者作為水包油液體乳劑或油包水液體乳劑而存在。活性成分還可作為丸劑、沖劑或糊劑存在。各種藥學上可接受的載體及其配方在標準配方論文中有述,例如,E.W.Martin的Remington’s Pharmaceutical Sciences。還參見Wang,Y.J.和Hanson,M.A.的Journal of Parenteral Science and Technology,Technical Report No.10,Supp.42:2S,1988。
片劑可任選與一種或多種助劑通過壓制或者模制制得。壓制的片劑可通過在合適的機器中對自由流動形式(如粉末或顆粒)的活性成分進行壓制而制備,所述活性成分任選地與粘合劑、潤滑劑、惰性稀釋劑、潤滑劑、表面活性劑或分散劑混合。模制的片劑可通過在合適的機器中對用惰性液體稀釋劑潤濕的粉末化合物的混合物進行模制而制得。片劑可任選地被包衣或者帶有刻痕(scored),并且可被配制,以便提供其中活性成分的緩慢或可控釋放。本化合物能夠以例如適用于立即釋放或者延長釋放的形式施用。立即釋放或者延長釋放能夠通過使用包含本化合物的合適的藥物組合物來實現。對于口服給藥,該化合物與遞送劑或載體進行配制,所述遞送劑或載體促進治療性大分子和高荷電的化合物穿過細胞膜(特別是在小腸中)進行轉運。此外,這樣的遞送劑或載體可抑制肽在通過胃腸(GI)道期間的酶降解,和/或該配方可包含另外的防止此類降解的藥劑。本化合物還能夠以脂質體進行施用。
用于口服給藥的示例性組合物包括可包含例如用于增量(imparting bulk)的微晶纖維素、作為懸浮劑的褐藻酸或褐藻酸鈉、作為增粘劑的甲基纖維素和本領域已知的甜味劑或調味劑的懸浮液;和可包含例如微晶纖維素、磷酸二鈣、淀粉、硬脂酸鎂和/或乳糖和/或本領域已知的其他賦形劑、粘合劑、增量劑、崩解劑、稀釋劑和潤滑劑的速釋片。發明的肽還能夠通過口腔以舌下含服或者口腔含化給藥進行遞送。模制片劑、壓制片劑或凍干片劑是其能夠使用的示例性方式。示例性組合物包括將本化合物與快速溶解稀釋劑如甘露醇、乳糖、蔗糖和/或環糊精進行配制的那些。這樣的配方中還包含高分子量賦形劑如纖維素(微晶粉末纖維素)或聚乙二醇(PEG)。這樣的配方還包含輔助粘膜粘附的的賦形劑,如羥丙基纖維素(HPC)、羥丙甲基纖維素(HPMC)、羧甲基纖維素鈉(SCMC)、馬來酸酐共聚物(例如Gantrez),以及控制釋放的試劑如聚丙烯酸共聚物(例如Carbopol 934)。還加入潤滑劑、助流劑、矯味劑、著色劑和穩定劑以便于制造和使用。
可包含的賦形劑為,例如,如人血清白蛋白的蛋白質或血漿制品。如果需要,藥物組合物還包含少量的無毒輔助物質,如濕潤劑或乳化劑、防腐劑、和pH緩沖劑等,例如乙酸鈉或失水山梨醇單月桂酸酯。
鼻腔噴霧或者吸入給藥的示例性組合物包括可包含例如芐醇或其他合適的防腐劑的鹽水溶液、增強生物利用度的吸收促進劑、和/或其他比如本領域已知的增溶劑或分散劑。方便地在鼻腔噴霧或吸入給藥的組合物中,借助合適的推進劑(例如二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷、二氧化碳或其他合適的氣體),發明的化合物從加壓包裝或者噴霧器中以氣溶膠噴霧的形式進行遞送。在加壓氣溶膠的情況下,劑量單位能夠通過提供閥門以遞送經計量的量來確定。用在吸入器或吹藥器中的例如明膠的膠囊和藥筒(cartridge)可配制為包含化合物和合適的粉末基質(例如乳糖或淀粉)的粉末混合物。在一個具體的非限制性實例中,發明的化合物作為噴霧劑通過也被稱為驅動器(actuator)的噴霧適配器從定量閥來進行施用。任選地,還包含穩定劑,和/或包含用于肺部深處遞送的多孔顆粒(例如,參見US專利號6,447,743)。
用于直腸給藥的配方可作為保留灌腸劑或者借助常用載體(如可可油、合成甘油酯或聚乙二醇)的栓劑存在。這樣的載體在常溫下通常為固體,但是在直腸腔內液化和/或溶解以釋放藥物。
用于在口中(例如向頰或舌下)的局部給藥的配方包括在調味基質(如蔗糖和阿拉伯樹膠或黃芪膠)中包含的活性成分的含片,以及在基質(如明膠和甘油或者蔗糖和阿拉伯樹膠)中包含的活性成分的錠劑。用于局部給藥的示例性組合物包含局部載體,如Plastibase(以聚乙烯凝膠化的礦物油)。
優選的單位劑量配方是包含活性成分的如上文詳述的有效劑量或者其合適分數的那些。
應當理解的是,除了上面具體提到的成分之外,考慮到所討論的配方的類型,本發明的配方包含本領域常規的其他試劑,例如適用于口服給藥的包含調味劑的那些。
發明的肽還適合作為持續釋放體系來施用。發明的持續釋放體系的合適實例包括適當的聚合材料,例如成型制品形式(例如膜或微膠囊)的半透型聚合物基質;適當的疏水材料,例如在可接受的油中作為乳液;或者離子交換樹脂;和發明化合物的微溶性衍生物,例如微溶性鹽。持續釋放體系通常經口服;直腸;腸胃外;腦池內(intracistemally);陰道內;腹腔內;例如作為粉末、軟膏、凝膠、滴劑或透皮貼劑的局部;向頰;或者作為口腔或鼻腔噴霧進行施用。
用于給藥的制劑可適當地配制成給發明的化合物帶來控制釋放。例如,藥物組合物為包含一種或多種生物降解聚合物、凝膠狀多糖和/或生物粘附性聚合物、兩親性聚合物、能夠改變發明的肽顆粒的界面性能的試劑的顆粒形式。這些組合物顯示出特定的生物相容性能,其允許活性物質的控制釋放。參見US專利號5,700,486。
發明的肽或藥物組合物的治療有效量能夠以單脈沖劑量、推注劑量或者隨時間推移施用的脈沖劑量進行施用。因此,在脈沖劑量中,提供發明的肽或組合物的推注給藥,隨著時間流逝接著進行第二次推注給藥。在具體的非限制性實例中,發明化合物的脈沖劑量在一天的過程中,在一周的過程中或在一個月的過程中施用。
具體實施方式
本發明是基于通過利用內源性調控機制經過控制緊密連接的動態開啟和關閉來打開上皮中相鄰細胞之間的細胞旁路的開發策略。該策略涉及提供靶向特異性細胞靶標并在對這些靶標具有特異性作用的全新(de novo)設計的肽化合物。
近來的研究已經表明,發生在一般粘膜表面(如腸道和氣道的那些)上的慢性炎癥事件與這些部位的上皮屏障透過性增加相關(4,13)。這種增加的透過性的基礎確定為緊密連接(TJ)相關的胞漿蛋白(被稱為肌球蛋白輕鏈(MLC))的磷酸化狀態的增加。這種機制通過選擇性抑制MLC磷酸化的九個氨基酸的透膜肽的識別和體內測試而被證實。MLC磷酸化的腸細胞調節的內源性機制的知識被用來設計兩類肽,以選擇性改變肌球蛋白磷酸酶靶亞基1(MYPT1)或17KDa L-激酶增效蛋白磷酸酶-1抑制劑(CPI-17)蛋白的功能。這兩種蛋白起到減少MLC磷酸化的作用,并且本發明涉及其功能的瞬時抑制,以瞬時提高上皮的細胞旁透過性。兩種靶蛋白以不同的速率、并通過提供選擇性地靶向這兩種不同的蛋白的試劑和方法來調控MLC磷酸化,最終,能夠控制緊密連接的開啟和再關閉的時間過程。
根據發明的第一方面,提供一種肽,其包含根據式1的序列:
X4-X5-X6-X7-X8
式1(SEQ ID NO:1)
其中:
X4為帶負電的氨基酸殘基,例如pTyr、Asp或Glu;
X5為小的疏水性氨基酸殘基,例如Val或Ala;
X6為帶正電的氨基酸殘基,例如Lys或Arg;
X7為親水性氨基酸殘基,例如Tyr、Phe、Thr或Ser;并且
X8為帶負電的氨基酸殘基,例如pTyr、Asp或Glu。
或者所述肽包含根據式2的序列的肽:
X5-X6-X7-X8
式2(SEQ ID NO:2)
其中:
X5為任意氨基酸殘基;
X6為Tyr;
X7為Gln;并且
X8為Tyr;
或者所述肽包含根據式1或式2的序列的逆-反形式,其中所有氨基酸殘基均為D-構型并且肽序列順序反轉。
式1限定了靶向PP1-CP1-17相互作用的序列。式2限定了靶向PP1-MYPT1相互作用的序列。兩種序列的一致特征在于它們都用于降低MLC磷酸化,因此它們的抑制導致MLC磷酸化和上皮的緊密連接打開的增加。
根據發明所有方面的一些實施方式,減少MLC磷酸化的功能性特征可為要求保護的發明的一個特征。
式1的序列
根據式1的肽抑制PP1-CP1-17相互作用。根據發明各個方面的一些實施方式,根據由式1所包含的序列,其中:
X4為pTyr、Asp或Glu;
X5為Val或Ala;
X6為Lys或Arg;
X7為Tyr、Phe、Thr或Ser;并且
X8為pTyr、Asp或Glu。
根據一些實施方式:
X4為Asp或Glu;
X5為Val或Ala;
X6為Lys或Arg;
X7為Tyr、Phe、Thr;并且
X8為Asp或Glu。
根據一些實施方式:
X4為Asp或Glu;
X5為Val;
X6為Lys;
X7為Tyr;并且
X8為Asp或Glu。
根據一些實施方式:
X4為Glu;
X5為Val;
X6為Lys;
X7為Tyr;并且
X8為Asp。
根據式1的各個實施方式的序列,限定了PP1-CP1-17靶向基序。為了在發明的一些實施方式中有效地起作用,包含根據式1的序列的肽還包含確保將其遞送進入上皮細胞的細胞液(其中具有其靶標)中的序列。這樣的序列為細胞透過-促進序列,或者在吞噬進入細胞之后促進該細胞表面與上皮細胞結合的序列。根據某個優選的實施方式,根據式1的序列為:
X1-X2-X3-X4-X5-X6-X7-X8-X9-X10(SEQ ID NO:5),或
X3-X4-X5-X6-X7-X8-X9-X10(SEQ ID NO:6),或
X1-X2-X3-X4-X5-X6-X7-X8(SEQ ID NO:7),
最優選X1-X2-X3-X4-X5-X6-X7-X8-X9-X10(SEQ ID NO:5)(式1a)
其中:
X1為至多10個帶正電的氨基酸殘基;
X2為帶正電的氨基酸殘基;
X3不存在,或者為疏水性氨基酸殘基;
X4為帶負電的氨基酸殘基;
X5為小的疏水性氨基酸殘基;
X6為帶正電的氨基酸殘基;
X7為疏水性氨基酸殘基;
X8為帶負電的氨基酸殘基;
X9為帶正電的氨基酸殘基;并且
X10為至多10個帶正電的氨基酸殘基;或者其中所有氨基酸殘基均為D-構型并且肽序列順序反轉。
作為涉及本發明的所有方面,根據上方給出的可供選擇的結構式的某些實施方式:
X1為至多10個帶正電的氨基酸殘基;
X2為帶正電的氨基酸殘基;
X3為Val、Ala、Leu或Ile;
X4為pTyr、Asp或Glu;
X5為Val或Arg;
X6為Lys或Arg;
X7為Tyr、Phe、Thr或Ser;
X8為pTyr、Asp或Glu;
X9為帶正電的氨基酸殘基;并且
X10為至多10個帶正電的氨基酸殘基;或者所有氨基酸殘基均為D-構型并且肽序列順序反轉。
根據一些實施方式:
X1為至多10個Lys或Arg殘基;
X2為Lys或Arg;
X3為Ala或Val;
X4為pTyr、Asp或Glu;
X5為Val或Arg;
X6為Lys或Arg;
X7為Tyr、Phe、Thr或Ser;
X8為pTyr、Asp或Glu;
X9為Lys或Arg;并且
X10為至多10個Lys或Arg殘基。
根據一些實施方式:
X1為Lys或Arg;
X2為Lys或Arg;
X3為Ala或Val;
X4為Asp或Glu;
X5為Val;
X6為Lys;
X7為Tyr;
X8為Asp或Glu;
X9為Lys或Arg;并且
X10為Lys或Arg。
根據一些實施方式:
X1為Lys或Arg;
X2為Lys或Arg;
X3為Val;
X4為Glu;
X5為Val;
X6為Lys;
X7為Tyr;
X8為Asp;
X9為Lys或Arg;并且
X10為Lys或Arg。
根據一些實施方式:
X1-X2-X3-X4-X5-X6-X7-X8-X9-X10為
Arg1-Arg2-Val3-Glu4-Val5-Lys6-Tyr7-Asp8-Arg9-Arg10(SEQ ID NO:3)
在某些實施方式中,發明還包含以上給出的序列的逆-反形式。
式2的序列
根據式2的肽抑制PP1-MYPT1相互作用。根據發明各個方面的一些實施方式,根據由式2所包含的序列,其中:
X5為帶正電的氨基酸殘基;
X6為Tyr;
X7為Gln;并且
X8為Tyr。
根據一些實施方式:
X5為Lys或Arg;
X6為Tyr;
X7為Gln;并且
X8為Tyr。
根據一些實施方式:
X5為Lys;
X6為Tyr;
X7為Gln;并且
X8為Tyr。
在某些實施方式中,發明還包含以上給出的序列的逆-反形式。
根據式2的各個實施方式的序列限定了PP1-MYPT1靶向基序。為了在發明的一些實施方式中有效地起作用,包含根據式2的序列的肽還包含確保將其遞送進入上皮細胞的細胞液(其中具有其靶標)中的序列。這樣的序列為細胞-透過-促進序列,或者在吞噬進入細胞之后促進該細胞表面與上皮細胞結合的序列。
根據某些優選的實施方式,根據式2的序列為:
X1-X2-X3-X4-X5-X6-X7-X8-X9-X10-X11-X12(SEQ ID NO:9),或
X1-X2-X3-X4-X5-X6-X7-X8-X9(SEQ ID NO:10),或
X3-X4-X5-X6-X7-X8-X9-X10-X11-X12(SEQ ID NO:11),
優選X1-X2-X3-X4-X5-X6-X7-X8-X9-X10-X11-X12(SEQ ID NO:9)
式2a
其中:
X1為至多10個帶正電的氨基酸殘基;
X2為帶正電的氨基酸殘基;
X3為Ala或Val;
X4不存在,或者為Ala,或者為1、2、3或4個帶正電的氨基酸殘基;
X5為帶正電的氨基酸殘基;
X6為Tyr;
X7為Gln;
X8為Tyr;
X9不存在,或者為Ala,或者為1、2、3或4個帶正電的氨基酸殘基;
X10為帶正電的氨基酸殘基;
X11為帶正電的氨基酸殘基;并且
X12為至多10個帶正電的氨基酸殘基;或者其中所有氨基酸殘基均為D-構型并且肽序列順序反轉。
涉及本發明的所有方面,根據上方給出的式2a的可供選擇的結構式的某些實施方式:
X1為至多10個Lys和/或Arg殘基的任意組合;
X2為Lys或Arg;
X3為Ala或Val;
X4不存在,或者為Ala、Lys或Arg;
X5為Lys或Arg;
X6為Tyr;
X7為Gln;
X8為Tyr;
X9為Ala或Lys或Arg;
X10為Lys或Arg;
X11為Lys或Arg;并且
X12為至多10個Lys和/或Arg殘基的任意組合;或者其中所有氨基酸殘基均為D-構型并且肽序列順序反轉。
根據一些實施方式:
X1為Lys或Arg;
X2為Lys或Arg;
X3為Ala;
X4不存在;
X5為Lys;
X6為Tyr;
X7為Gln;
X8為Tyr;
X9不存在;
X10為Lys或Arg;
X11為Lys或Arg;并且
X12為Lys或Arg。
根據一些實施方式:
X1為Arg;
X2為Lys;
X3為Ala;
X4不存在;
X5為Lys;
X6為Tyr;
X7為Gln;
X8為Tyr;
X9不存在;
X10為Arg;
X11為Arg;并且
X12為Lys;
也就是說,序列為Arg1-Lys2-Ala3-Lys5-Tyr6-Gln7-Tyr8-Arg10-Arg11-Lys12(SEQ ID NO:4).
在某些實施方式中,發明還包含以上給出的序列的逆-反形式。
逆-反(retro-inverso)序列
式1和式2的序列衍生自天然存在的序列。衍生出它們的天然存在的序列具有L-構型的氨基酸殘基。因此,在這樣的情況下,根據一些實施方式,氨基酸是L-構型的。
包含D-型氨基酸的肽作為藥物更具吸引力,因為它們傾向于更少受到胃內或細胞內部由于蛋白水解而降解的影響。因此,它們傾向于更加適用于口服,并且傾向于在更長的時間內更有效。
本發明在其各個方面包括其中所有氨基酸均為D-構型并且式1和2(以及落入式1或2和如上詳述的范圍內的更窄限定的序列)的肽序列順序反轉的肽。
例如,發明涉及具有殘基優選為L-構型的序列RRVEVKYDRR-NH2(SEQ ID NO:3)的肽,和具有殘基為D-構型的逆-反的序列NH2-rrdykvevrr(SEQ ID NO:20)的肽。小寫的單字母氨基酸碼是逆-反構型的簡寫。根據某些實施方式,逆-反構型是優選的。
藥物組合物
根據發明的第二方面,提供有包含任意根據發明第一方面的各個實施方式的肽和藥學上可接受的載體的藥物組合物。
根據某些實施方式,藥物組合物包含促進肽透過進入靶上皮細胞的細胞液的試劑,例如脂質體形成劑。如果肽本身缺乏細胞透過序列,例如如果其缺乏式1a或2a的細胞透過肽序列,這樣的試劑的存在是特別受青睞的。然而,根據一些實施方式,無論在根據發明第一方面的肽中這樣的序列存在或者不存在,都存在輔助細胞透過的試劑。根據某些實施方式,根據發明的藥物組合物包含根據式1的肽或者其逆-反類似物,與根據式2的肽或者其逆-反類似物的結合。
腸溶衣
藥物組合物包括具有腸溶衣的劑型。腸溶衣為應用至口服藥物組合物(如片劑、囊片和膠囊)以便控制吸收使得吸收在小腸發生的包衣。腸溶衣通常應用到劑型表面上,由此其在胃的高酸性pH中存在穩定的表面,但是在小腸相對更堿性的環境中迅速崩解。用于腸溶衣的材料包括脂肪酸、石蠟、塑料和植物纖維。
當本發明用于上下文中將口服劑量的第二治療劑遞送穿過小腸上皮時,如果不然治療劑會在胃環境中降解,則可使用腸溶衣。如果第二治療劑對胃有刺激、如果其是酸不穩定的(例如某些唑類藥物如艾美拉唑是酸不穩定的)、或者如果第二治療劑為預計會被胃中存在的酶降解的蛋白質或肽(例如胰島素、GLD-1或其衍生物或類似物),這樣的包衣可能特別有用。
因此,根據發明第二方面的藥物組合物包括具有腸溶衣的固體劑型,并且發明的其他方面還利用或涉及這樣的包衣。
第二治療劑
根據涉及發明的肽和第二治療劑的發明的各個方面,例如,根據發明第二方面的藥物組合物,兩種組分優選例如在藥物組合物中共混,由此它們在相同時間存在于上皮表面處。發明的肽可用于遞送廣泛的治療劑。特別地,它們可與小分子治療劑的遞送相關,并且還與肽或蛋白質或核酸的治療劑的遞送相關。例如,治療劑為胰島素、甲狀旁腺激素、降鈣素、促紅細胞生成素、GM-SF或生長激素、或者siRNA或基因治療劑。發明還涉及抗體或抗體衍生物的治療劑的遞送。應當發現,全抗體分子太大以至于無法根據發明被有效遞送,基于抗體技術的更小變體,例如scFv分子、駱駝抗體(camel antibody)、單變化抗體片段和結構域等可能特別有用。在通過發明的肽打開緊密連接之后,所有這些治療劑能夠單獨地或者與其他治療劑聯合施用。例如,胰島素和GLP-1能夠以與發明的肽相同的劑型被遞送。
藥學上載體
藥學上載體(或“藥物載體”)為藥物組合物中存在的改善藥物的遞送、配方、劑量或療效或輔助其保存、混合、制備或加工的物質。
上皮
發明在各個方面涉及上皮(“上皮表面”)。這可能是體外或體內的表面。非限制性的實例包括存在于體內和體外二者中的大腸上皮、小腸上皮、直腸上皮、肛門上皮、宮頸上皮、陰道上皮、呼吸道(例如氣管、細支氣管或喉部)上皮、鼻上皮、口部上皮和眼角膜上皮。
特別考慮到了上皮細胞(例如以上所述的上皮細胞類型)的永生化細胞系。
藥物
根據發明的第三方面,提供有根據發明第一方面的肽或者根據發明第二方面的藥物組合物,其用作藥物。發明的第三和第四方面都是指“藥物”。參考發明的其他方面,所述藥物包含本文所述的發明的肽或者發明的肽和第二治療劑。該第二治療劑為肽、小分子實體、核酸或其他化學組成(chemical compound)。藥物如本文所述參考發明的其他方面被配制,用于治療本文其他部分所述的受試者的本文其他部分所述的疾病病癥或非醫學情況。
用于治療的受試者
根據本發明的某些實施方式,在所有方面均涉及受試者的治療。這樣的受試者為具有緊密連接的上皮的任何物種。根據某些優選的實施方式,本發明在所有方面均涉及哺乳動物受試者的治療。最優選地,本發明在所有方面均涉及任一性別在任何年齡(例如從出生至1歲、出生至5歲、出生至12歲、出生至60歲、出生至80歲、2至60歲、5至80歲、12至80歲、或者16歲或18歲以上)的人受試者的治療。
根據某些實施方式,本發明在所有方面均涉及需要將治療劑穿過粘膜進行遞送的人受試者的治療。治療劑為本文其他部分所述藥劑的一種。根據發明所有方面的某些實施方式,人受試者為在已經向其施用上述治療劑的同時或在15、10、5、2或1分鐘的時間窗口內任一方面施用發明的肽或藥物組合物的受試者。
治療的疾病
根據第一方面的肽,根據第二方面的藥物組合物,或者根據第三方面的藥物用于治療疾病或者病癥或者其他病情。根據某些實施方式,疾病或肺病,根據本發明的吸收較差的藥物的遞送可能具有與通過注射給藥類似的效果。重要的是,將治療劑遞送至分散的上皮部位的能力還將引起粘膜下空間的局部遞送。出于這些原因,發明特別適用于治療全身性病情和疾病(例如,關節炎、身材矮小等)以及將得益于治療劑的局部靶向的病情和疾病(例如,克羅恩病、哮喘等)。
非治療性治療
根據某些實施方式,發明的各個方面涉及病情(如化妝品皮膚病)的非治療性治療,或者治療并非醫學性肥胖但為了美容目的想要減輕體重(或者保持健康體重)的人。
藥物治療方法
考慮到構成可授權主題的藥物治療方法的權限,本發明提供治療受試者的疾病的方法,其包括向所述受試者施用根據發明第二方面的藥物組合物,其中第二治療劑用于治療疾病,并且其中藥物組合物中存在的根據發明第一方面的肽改善了第二治療劑穿過受試者上皮的遞送。根據發明這一方面的某些優選實施方式,參考發明的其他方面,受試者、給藥速率、藥物組合物的特點、發明的肽、第二治療劑、或者要治療的疾病為如本文所述。
打開緊密連接的方法
根據發明的第四方面,提供有打開上皮表面的緊密連接的方法,其包括將有效量的根據發明第一方面的肽,或者根據發明第二方面的藥物組合物施用于上皮。
該方法為體內或體外方法。體內方法包括針對受試者的藥物治療方法,例如,通過借助打開所述受試者上皮的細胞旁給藥速率而促進第二治療劑向受試者的血液循環遞送,并且通過參考發明的其他方面,要治療的疾病為如本文所述。
或者,發明這一方面的方法涉及在實驗動物或人類志愿者或患者中遞送藥劑穿過上皮的體內試驗。例如,為了測量第二治療劑在治療疾病中的療效,或者為了輔助隨后的藥代動力學(PK)參數的測量,通過參考發明其他方面所限定的第二治療劑可穿過上皮進行遞送。就此而言,發明這一方面的方法涉及穿過所述上皮的標志物分子(例如放射性標記的標志物或其他顯像劑或造影劑)的測量。
或者,為了研究上皮或者穿過所述上皮的分子的通道的性質,發明的這一方面涉及在細胞培養物(例如人類或動物上皮的單層培養的細胞)中的上皮的緊密連接的體內開啟。
遞送方法
根據發明的第六方面,提供有遞送藥劑穿過上皮表面的方法,其包括將所述藥劑與根據發明第一方面的肽聯合施用,或者作為根據發明第二方面的藥物組合物的一部分進行施用。
在發明這一方面的各個實施方式中,上皮表面、肽和藥物組合物為如參考發明其他各個方面所述。該方法可在體外或體內進行。藥劑為診斷劑、造影劑或者如本文所述在“第二治療劑”標題下的根據某些優選實施方式的治療劑。
本發明的各個方面在下文參考以下的非限制性實例和附圖進行描述。
實施例
材料和方法
肽合成
使用從Nova Biochem獲得的氨基酸(除異亮氨酸從Sigma Aldrich獲得之外)通過(Fmoc)-SPSS(ActivoSyn肽合成器)合成肽。用N,N’-二異丙基碳二亞胺將第一個氨基酸偶聯到Rink Amide MBHA樹脂(100-200目;Nova Biochem)。隨后使用PyBop在Activo P-11肽合成器上進行偶聯。在二甲基甲酰胺中用20%的哌啶進行脫保護。用三氟乙酸(TFA)、三異丙基硅烷和水(95:2.5:2.5)的混合物將肽從樹脂中分開,然后在二乙醚中沉淀。使用Phenomenex Gemini C18柱(250x10mm,孔徑5μm)以及具有0.1%TFA的水和具有0.1%TFA的乙腈的梯度流動相、用2.5ml/min的流速,通過HPLC對粗產物進行純化。在Bruker Daltonics的micrOTOF質譜儀上用噴霧電離質譜(ESI)獲得高分辨率飛行時間質譜,以驗證肽的同一性。通過凍干將純化的肽干燥并儲存在-80℃。
細胞培養物
將永生化的人腸上皮細胞系(Caco-2)保持在補充有10%的FBS、2mM的L-谷氨酰胺(Gibco)、100U/ml的青霉素和100μg/ml的鏈霉素(Gibco)的DMEM/F12培養基(Gibco,Paisley,UK)中。將Caco-2細胞以7x104/孔的密度接種在TranswellTM(Corning,NY)聚酯膜濾器上(直徑12mm,孔徑0.4μm)。每兩天用DMEM(Life Technologies,Paisley,UK)換液。具有>350Ω·cm2的跨上皮電阻(TEER)的Caco-2單層被認為是鋪滿的,并被用在轉運試驗中;固定式電極用于測量TEER(World Precision Instruments,UK)。
體外轉運研究
進行50mg/ml 4kDa的葡聚糖或50mg/ml 70kDa的葡聚糖(Sigma)的頂部至基底的通量以評估PIP肽對細胞旁透過性的影響。頂部隔室(compartment)體積為200μl;以設定的次數收集到600μl的基底隔室體積并用新鮮的HBSS替換。用Floustar Omega酶標儀(BMG Labtech,Ortenburg,德國)測定頂部和基底的隔室的熒光。在HBSS替換之后立即對每個孔進行TEER測量。3小時后,除去頂部隔室的葡聚糖和肽溶液,并用PBS替換,進一步記錄30分鐘的TEER值以評估單層的恢復。
磷酸化MLC分析
在用冰冷的PBS沖洗之后,將Caco-2單層或者來自分離的腸組織的上皮細胞在3μL的蛋白酶抑制劑混合物和200μL RIPA緩沖液中裂解,并在冰上放置10分鐘。將分離的腸組織類似地置于冰冷的PBS中15分鐘,然后在蛋白酶抑制劑混合物和RIPA緩沖液中浸漬,在冰上放置10分鐘。將所有細胞和組織裂解物在8000rpm下離心15分鐘以收集上清液,將其貯存在-80℃直到使用。對于蛋白質印跡分析,通過在220V下運行40分鐘的SDS-PAGE(12%)對裂解物進行分離,并且用XCellTM Blot模塊(Invitrogen)在30V下電轉移至PDVF膜上70分鐘。在TBS-T(2M Tris HCl,pH 7.5;4M NaCl和0.1%的吐溫20)中用5%的牛血清白蛋白將膜封閉1小時。將膜用水清洗,與第一抗體(抗肌球蛋白輕鏈(磷酸化S20)抗體或者抗肌球蛋白輕鏈2抗體)在5℃溫育過夜,用TBS-T清洗三次,然后與辣根過氧化物酶(HRP)偶聯的第二抗體在室溫下溫育1小時。在TBS-T中清洗三次之后,通過ECL(Santa Cruz)檢測HRP的活性。
細胞活力測定
MTT試驗:用3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四氮唑溴(MTT;Invitrogen,Paisley,UK)試驗來評估細胞活力。在將細胞暴露于CPP復合物72小時之后,將0.5mg/ml的MTT加入培養基并與細胞進一步孵育3小時。然后將培養基除去并用DMSO替換以使細胞裂解。然后記錄在550nm處的吸光度。
顯微鏡檢查
在加入CPP之前,將細胞在玻璃蓋玻片上培養,與羅丹明(Rhodamine)標記的BSA復合。將細胞與復合物孵育2小時,之后接著用完全培養基洗滌數次以除去過量的復合物,然后以4%的多聚甲醛固定。通過共聚焦顯微鏡(Zeiss LSM 510)測定標記的BSA引入細胞中。BSA進入的水平通過測定相對于視野中細胞數目的羅丹明標記的平均熒光強度來估算。
體內研究
當進行研究時,內部培育的雄性Wistar大鼠為225-275g(大約6-8周大小)。將大鼠以每籠3-5只一組、12小時光照/12小時黑暗一周期的方式進行安置。所有實驗均在光照階段進行,并且使用連續異氟醚麻醉的無法恢復的實驗方案來實施。進行4-5cm的腹部正中切口以暴露空腸中段至小腸的早期回腸區域,并為套管的插入提供門靜脈入口。在包含10mM檸檬酸的0.9%的鹽水中制備胰島素(人類重組;Sigma)和PIP肽的溶液以減少局部蛋白水解,并以1:1進行混合,然后用29號注射針以200μL/kg的體積(或者每250g大鼠約50μL)進行注射。用永久性記號筆對注射部位進行標記。用外科膠布封住切口。在接下來的兩個小時從門靜脈以及體循環抽血以監測血糖、血清胰島素和內毒素。對所有組使用的對照治療組包括:將胰島素遞送進入腸腔的SC注射,以及不具有胰島素或者胰島素單獨在磷酸鹽緩沖液(PBS)配方中的PIP肽的腸道注射。在研究結束時,將大約3-5mm的標記的腸片段區域分離。在經執業的獸醫病理學家分析解讀之前,將組織裂解用于MLC磷酸化狀態的生物化學評估或固定、切片,并用蘇木精/伊紅進行染色。在肩胛骨中間區域進行皮下(SC)胰島素注射(20μl/kg),并以相同的方式測量血糖。所有實驗根據1986年的英國動物(科學程序(Scientific Procedures))法、1986年的歐共體理事會指令(European Communities Council Directive)(86/609/EEC)和巴比倫(Bath)大學的倫理審查程序而進行。
體內毒性試驗
由于細胞透過性肽的作用有可能誘發細胞損傷和毒性(Carter,2013#109;Kilk,2009#110),通過使用APT165商品化試劑盒根據每個制造商的說明書(Millipore,Watford,UK)檢測半胱天冬酶-3的酶活性來評估作為早期細胞中毒測量的細胞凋亡的誘發。在暴露于通過直接腸腔內注射而施用的檢測藥劑45分鐘之后,將腸組織分離。將潮霉素(150μg/mL)作為正對照施用以通過半胱天冬酶-3活化來促使細胞凋亡。
磷酸化MLC分析
在用冰冷的PBS沖洗以除去PIP肽或對照治療劑之后,將分離的腸組織在冰冷的PBS中放置15分鐘,然后加入25μl蛋白酶抑制劑混合物(Fisher)、25μl磷酸酶抑制劑混合物(Fisher)和500μl RIPA緩沖液(Sigma Aldrich)。在冰上10分鐘之后,將裂解物在8000rpm下離心15分鐘以收集上清液,將其貯存在-80℃直到使用。對于蛋白質印跡分析,通過在220V下運行40分鐘的SDS-PAGE(12%)對裂解物進行分離,并且用XCellTM Blot模塊(Invitrogen)在30V下電轉移至PDVF膜上70分鐘。在TBS-T(2M Tris HCl,pH 7.5;4M NaCl和0.1%的吐溫20)中用5%的牛血清白蛋白將膜封閉1小時。將膜用水清洗,與第一抗體(抗肌球蛋白輕鏈(磷酸化S19)抗體(Cell Signalling Technologies)或者抗肌球蛋白輕鏈2抗體(Abcam))在5℃溫育過夜,用TBS-T清洗三次,然后與辣根過氧化物酶(HRP)偶聯的第二抗體在室溫下溫育1小時。在TBS-T中清洗三次之后,通過ECL(Santa Cruz)檢測HRP的活性。
細胞活力測定
通過使用APT165商品化試劑盒根據每個制造商的說明書(Millipore,Watford,UK)檢測半胱天冬酶-3的酶活性來評估作為早期細胞中毒測量的細胞凋亡的誘發。在暴露于通過直接腸腔內注射而施用的檢測藥劑45分鐘之后,將腸組織分離。將潮霉素(150μg/mL)作為正對照施用以通過半胱天冬酶-3活化來促使細胞凋亡。
顯微鏡檢查
對于顯微分析,在給藥后15分鐘將體內暴露的腸段分離,在此對PIP肽介導的Cy3-標記的胰島素(Nancocs)的攝取進行評估。分離的組織簡單地在冰冷的PBS中沖洗,然后用4%的多聚甲醛固定,之后用Zeiss LSM 510熒光顯微鏡進行評估。DAPI(4',6-二脒基-2-苯基吲哚)用作核染色劑。
數據分析
通過扣除空白濾波器的讀數并對單層而言標準化為初始TEER值的百分比來計算TEER值。如前所述進行用于計算細胞旁透過性的熒光葡聚糖通量。對每個動物而言血糖水平表示為基線葡萄糖水平的百分比。使用雙尾未配對Student氏T檢驗(two-tailed unpaired Student’s T-test)將數據與對照值進行比較。p值<0.05被認為顯示出顯著的差異。
實施例1.緊密連接(TJ)結構和功能的動態控制的背景理論
TJ結構由內在膜蛋白(integral membrane protein)構成,所述內在膜蛋白與可收縮及骨架成分相互作用以在健康或疾病中動態地調控上皮屏障性質(Turner(2009)Nat.Rev.Immunol.9:799-809;Xiao等,(2011)J.Aller.Clin.Immunol.128:549-556)。生理學和病理學刺激通過多種激酶(蛋白激酶C(PKC)、蛋白激酶A、絲裂原活化蛋白激酶、磷酸肌醇3-激酶和Rho信號通路)改變TJ屏障性質(Gonzalez-Mariscal(2008)Biochim.Biophys.Acta 1778:729-756)。這種激酶交叉感知(cross-talk)的核心要素涉及調節肌球蛋白輕鏈(MLC)磷酸化(Woodsome等,(2006)J.Cell Sci.119:1769-1780);圖1,增加的MLC磷酸化和降低的TJ屏障功能之間存在直接的聯系;借助起關鍵作用的MLC激酶(MLCK),促炎性細胞因子可以驅動MLC磷酸化,導致TJ的開啟。發明人在先已經確認了一種膜透過的穩定的MLCK肽抑制劑,被稱為PIK(圖1中箭頭),其能夠糾正腸和肺中炎癥驅動的TJ屏障功能降低(Clayburgh等,(2005)J.Clin.Invest.115:2702-2715;Mirzapoiazava等,(2011)Am.J.Respir.Cell Molec.Biol.44:40-52)。據推測MLCK的功能被MLC磷酸酶的作用所抵消。雖然對這樣的網絡中的靶標使用基因敲除的研究可以提供對其在屏障功能中作用的洞察,但是通過針對這樣的網絡中單一靶標的任何分子對整個體系的作用必須通過藥理學研究來確定。即,這些蛋白質表面接觸位點是細胞內的,并且這些藥劑的相關試驗的第一位點為腸,成功的抑制劑應當優選兼有穩定性和膜可透過性。
實施例2.初步研究
發明人在先已經開發了MLCK的9個殘基的肽(PIK)抑制劑,其封閉由于炎癥事件而處于開放位置的TJ結構。PIK模仿了對MLCK功能而言關鍵的蛋白質表面結構。在確認了關鍵的界面接觸之后,修飾該肽以整合氨基酸,這將允許細胞透過性肽(CPP)起作用并增加代謝穩定性。采用這些相同的設計理念,已經開發出根據本發明各個方面的試劑和方法。
MLCP為三聚蛋白磷酸酶-1(PP1)全酶,其由PP1異構體、肌球蛋白靶向亞基MYPT1-CPI-17調節復合物以及21kDa輔助亞基構成。為確認潛在的肽MLCP抑制劑,發明人已經關注MYPT1或17kDa蛋白抑制劑CPI-17與PP1之間的相互作用,以直接地調控MLCP功能(圖3)。
實施例3.靶向PP1-CPI-17相互作用
通過CPI-17的MLCP抑制由PKC刺激的事件驅動;CPI-17中的殘基T38的磷酸化(pT38)使MLCP的抑制作用增強了超過1000倍;確定了在CPI-17中的最小抑制結構域,其包括T38(24),并且合成了模仿CPI-17中的R36VTVKYDRR44(SEQ ID NO:13)序列的肽的小骨架,所述R36VTVKYDRR44序列參與其與PP1的相互作用。初始肽組中包括模仿pT38(谷氨酸;E)的修飾,并且引入另外的要素(basic)以模仿CPP序列,以增強膜透過性(表1)。
表1顯示出測試的CPI-17衍生肽的結果。選擇R36VTVKYDRR44(SEQ ID NO:8)作為起始序列(條目1)。在測試系列(條目2-6)中殘基如所示的進行變化(黑體,下劃線)。在頂部應用到Caco-2細胞單層之后,功效通過TEER降低來測定。
將肽以5mM的濃度頂部應用至在半透的插入小室(insert)上生長的Caco-2細胞鋪滿的單層上(Rubus等,(1996)J.Pharma.Sci 85:165-169),以評估其影響TJ功能的能力(Peterson&Mooseker(1992)J.Cell.Sci.102(3):581-600)。在加入25分鐘之內,RRVEVKYDRR(6#肽;肽640,SEQ ID NO:3)使Caco-2單層的跨上皮電阻(TEER)降低50%(圖4),并且根據MTT試驗,這些肽在該濃度下沒有一種是有毒性的(Mosman(1983)J.Immunol.Methods 65:55-63)。關鍵是,在溫和清洗之后,RRVEVKYDRR(SEQ ID NO:3)治療過的Caco-2單層TEER值在24小時之內恢復到其初始值的80%。基于RRVEVKYDRR(SEQ ID NO:3)序列確認了首選的侯選物:肽NH2-rrdykvevrr-NH2(SEQ ID NO:20),但是使用具有D-構型氨基酸的逆-反構型。
雖然CPI-17在平滑肌中廣泛表達(Eto(2009)J.Bid.Chem.369:149-156),但是該數據是第一次提出其在分化的上皮細胞中的功能性作用。雖然pT38對CPI-17本身的抑制性功能必不可少,但是本發現指明一種合適的穩定模擬物(例如,E殘基)能夠替代pT38用于我們的應用。表1的肽的N和C末端作為細胞透過性序列而提供,但是如果采用其他實現細胞透過性的方法也可以不需要它。
實施例4.靶向PP1-MYPT1相互作用
在Rho-激酶介導的MYPT1在殘基T696和T853處的磷酸化之后發生MLCP抑制(Murthy等,(2003)Biochem.J.374:145-155)。MYPT1通過三個不同的區域與PP1結合:RVxF結合基序、N-末端臂和該蛋白的第二組錨蛋白重復序列。雖然MYPT1在T696和T853二者處的磷酸化似乎參與MLCP調控,但是在MYPT1的300個殘基N-末端結構域中的KVKF序列促進其與PP1的聯合(Peti等,(2013)FEB S.J.280:596-611)。MYPT結合至PP1的結構分析顯示PP1的E300至E309位于MYPT的兩個錨蛋白重復序列之間;特別是,錨蛋白重復序列與Y305和Y307結合,表明PP1的C末端對介導異構體特異性的調節亞基相互作用是非常重要的。因此,推測與該區域對應的肽能夠防止MYPT結合至PP1,并由此減少MYPT/PP1復合物的肌球蛋白特異性。
為測試該假說,合成了包含PP1的C末端區域的301KAKYQY306(SEQ ID NO:19)區域的RKAKYQYRRK(肽250,SEQ ID NO:4)。靶向的面間接觸的評估表明可以分別在N和C末端加入R殘基和三肽RRK,而不干擾促進CPP起作用的功能。由于聚陽離子可引發細胞損傷和毒性(Wender等,(2005)Org.Lett.7:4815-4818),用MTT試驗確認對該肽來說缺乏細胞毒性(數據未顯示)。將5mM肽250頂部添加至分化的Caco-2單層與添加5mM RRVEVKYDRR(6#肽;肽640,SEQ ID NO:3)以及對照肽進行比較(圖5)。雖然相對于對照,肽250和640均增加了細胞旁標志物的轉運,但是它們表現為不同的時間過程并且對TEER具有不同的影響。發明人選擇4kDa的熒光葡聚糖(4FD)作為細胞旁標志物,因為該分子的流體動力學半徑略大于胰島素和GLP-1的,因此應當預示它們的轉運。靶向PP1-CPI-17相互作用的肽640,引起TEER迅速降低至其初始值的約50%,并且幾乎立即引起了4FD轉運的增加;這些作用與在第45分鐘時洗掉并在第70分鐘時再次加入的肽密切偶聯。與肽640相比,靶向PP1-MYPT1相互作用的肽250,產生了較慢且較不顯著的TEER降低,以及較不顯著的4FD轉運中的初始增加。有趣的是,雖然在第45分鐘時除去肽250引起TEER的恢復,但是在第70分鐘時再次加入肽無法影響TEER,而是進一步增強了4FD的轉運。
基于以上信息,確定了首選的侯選物為肽250:NH2-krryqykakr-NH2(SEQ ID NO:21),其使用逆-反構型和D-型氨基酸。
這些研究顯示,就TEER和細胞旁轉運而言,為阻斷CPI-17或MYPT1與PP1的相互作用而合理設計的肽能夠產生獨特的藥理學成果,表明存在包括CPI-17和MYPT1在內的獨立的調節機理,這允許TJ動態開啟至各種程度和速度。
體外研究
為了充分地對PIP肽250和640進行比較,我們嘗試基于對以TEER為基礎的響應將二者進行匹配。這樣做是因為幾種不確定因素使得直接劑量比較不適當,因為可能兩種肽具有對它們的作用關鍵的不同性質:對其細胞內靶標的可及性(由于CPP能力不同)、對其各自的MLCP-相關的靶標的親合力、脫靶(off-target)作用和細胞內穩定性。我們確定20mM肽250提供了與10mM的肽640相當的響應(圖6)。減少兩種PIP肽的頂部劑量表明,各自誘導TEER中的劑量依賴性變化,包括起始速率和最大效應(圖6)。在PIP肽暴露180分鐘后,將新鮮培養基替換至頂部隔室顯示,這些PIP肽的除去引發TEER抑制的快速逆轉,暴露于肽640的細胞可能比那些暴露于肽250的細胞更快地響應(圖6)。這樣的觀察表明,由肽250誘導的細胞改變的恢復可能不如肽640作用的通路活躍。由于肽640設計為影響PKC-介導的MLCP調節子,并且肽250設計為影響Rho激酶-介導的MLCP的調節,PKC和Rho-激酶在TJ結構/功能的快速和更持久的改變中所發揮的覺察到的(perceived)作用一致。
接下來我們詢問PIP肽640和250對TEER的作用是否轉化為細胞旁透過性的變化(圖7)。盡管在暴露180分鐘之后在TEER中產生了接近50%的損失,但是5mM的肽640無法影響4kDa葡聚糖穿過Caco-2單層的透過性。10mM肽640的頂部施加使4kDa透過性的速率增加了3倍。有趣的是,就4kDa通量率而言,10mM肽640等同于10mM肽250,盡管與其他肽相比對TEER變化具有略微延遲和較弱的作用。令人驚訝的是,與10mM肽640相比,即使TEER變化圖對這些治療而言幾乎是相同的,但20mM肽250導致對4kDa葡聚糖而言通量率加倍(與對照相比大約6倍)。我們還觀察到肽640無法影響70kDa葡聚糖的通量,而20mM下的肽250可以使該通量增加約3倍。這些增強的4kDa葡聚糖通量的線性度(圖7)表明,不管達到TEER響應平臺的時間,由這些PIP肽引起的透過性改變的作用非常迅速(圖6)。有跡象表明由20mM肽250誘導的增強的70kDa葡聚糖轉運的誘導有輕微的延遲。總的來說,這些結果表明,與由肽250誘導的等價作用相比(基于TEER),肽640誘導更動態和不太強鍵的分子旁路徑的開啟。
體內研究
我們的焦點在于在解決與繞開胃酸和胰腺酶相關的配方挑戰之前檢查腸上皮轉運;這樣的挑戰可以借助已建立的藥丸或片劑技術來解決,但是需要比嚙齒動物更高級的動物模型。目前,我們使用大鼠模型,其中將較小體積(50μL)直接注入小腸腔中,具體為遠端空腸至近端回腸。我們通過胰島素的皮下(SC)注射校正通過口服遞送達到的響應;劑量依賴性的血糖降低在約30分鐘達到最低點并且在約90分鐘時開始恢復。3IU/kg的SC注射產生約50%的血糖降低(圖8a)。將30IU/kg直接腔內注射至大鼠的小腸(空腸中段至回腸中段)對血糖沒有影響(圖8b)。然而,30IU/kg的胰島素加上10mM的肽250的ILI引起與SC注射3IU胰島素所觀察到的類似的血糖降低和恢復曲線,注意到在90分鐘時已經達到了70-80%的血糖恢復(圖8a、b)。有趣的是,通過ILI以30IU/kg施用的20mM的肽250引起30分鐘時40%的血糖降低,在試驗的剩余階段保持在這一水平(圖8b)。與SC注射(圖8a)或10mM肽250(圖8b)相比,30IU/kg的胰島素與10mM肽640的直接ILI引起血糖變化曲線中降低大大延遲(圖8c);需要20mM肽640的ILI以產生與用10mM的肽250所觀察到的類似的效果。
這些結果是有趣的,因為它們表明這些PIP肽在體外和體內具有不同的響應曲線,這可能與其響應的耐久性有關。在體外,采用的PIP肽保持存在和功能直到其被物理地除去。然而,在體內,在腔上皮表面處的PIP肽的有效濃度通過某些事件如腔沖洗和全身吸收而復雜化;可能改變PIP肽的量和持續時間的事件可能在小腸上皮細胞的細胞質中。我們觀察到,肽640對TEER顯現出比肽250更動態的響應,這會引起體內更持久的響應。
PIP肽作用的機理
這些體內結果通常與我們用人腸上皮細胞系Caco-2進行的體外研究相一致,其表明細胞旁透過性的動態變化(圖6和7)。如所預期的,這些PIP肽在體內的作用是瞬時的,并且它們作用的持續時間和起始時間是劑量依賴性的。PIP肽250和640在體外的區別是顯而易見的,其中它們的絕對濃度和暴露持續時間是可控的。通過將MLC的位置19處的磷酸絲氨酸(pMLC)的程度與從經血糖測定的時間過程的PIP肽暴露部位分離出的大鼠腸組織中的總MLC進行比較來測定MLC的磷酸化狀態,我們檢查了PIP肽250和640在體內作用的起始和持續時間(圖8B和8C)。
通過半定量蛋白質印記分析(圖9A)來評估總MLC與pMLC之比。該分析顯示,由20mM肽640誘導的MLC磷酸化比例在15分鐘內顯著增加,并在45分鐘時保持升高,然后在90分鐘時回到初始水平之前。20mM肽250達到的MLC對pMLC比例曲線在15分鐘時顯示無明顯的變化,而在45分鐘時升高,然后在90分鐘時回到基底水平(圖9B)。這些結果與由這些PIP肽和胰島素的共同給藥誘導的血糖降低的時間過程非常相關(圖8B和8C)。重要的是我們注意到,當關注僅從腸上皮細胞中分離的MLC時,使用的提取過程可能已經引起這些分離的腸組織中存在一些從其他細胞分離的MLC。
通過檢查在其ILI注射之后胰島素的熒光標記形式的去向,我們進一步驗證了PIP肽作用機理的假說。只有當與PIP肽共同施用而沒有上皮的大體解剖修飾時,在腸細胞旁空間觀察到熒光(cy3-標記)胰島素(圖9C)。這些結構共同地支持了該假說,即,PIP肽的頂部、局部施用起到局部增加體內大鼠腸上皮的細胞旁透過性的作用。進而,胰島素的細胞旁透過性增加的時間過程與上皮細胞中pMLC含量的瞬時增加的作用是一致的。
PIP增強的胰島素攝取的去向
在ILI注射PIP肽和10IU胰島素之后,監控血糖降低和總的MLC細胞含量有關的MLC磷酸化的變化的時間過程研究表明了大約30-60分鐘的體內事件窗口期。為進一步探究,我們測量了在ILI之后5到90分鐘從門靜脈(圖10A)和尾靜脈(圖10B)收集的血液中的血清胰島素濃度。門靜脈中胰島素水平增加的起始時間和這些增加的水平的持續時間表明,與肽250相比,肽640激起了更迅速的作用起始時間和更短的作用持續時間,這與這兩種肽的磷酸化比例改變來說的時間過程是一致的(圖9)。
在10IU/kg的ILI之后,通過在門靜脈和全身(尾靜脈)中的ELISA檢測的總胰島素量,對于本文的試驗條件,在通過肽250相比肽640增強攝取之后顯示略微不同的曲線。另外,我們測定了SC注射之后胰島素的門靜脈和全身濃度的時間-濃度曲線(圖10C)。采用非小室模型(non-compartmental)分析和Wagner-Nelson去卷積(deconvolution),當通過ILI施用肽250和肽640時,對于在門靜脈中檢測出的人胰島素而言,口服給藥(相對于SC)的相對生物利用度分別為4%和3%。有趣的是,在ILI施用肽250和肽640之后,對于到達體循環的人胰島素而言,相對生物利用度分別為1.6%和1.4%。這些結果表明相當一部分遞送至門靜脈的人胰島素沒有到達體循環。
在PIP肽暴露之后的上皮細胞活力
采用Caco-2細胞的初始體外研究表明,在調節TEER和細胞旁透過性的濃度下測試的肽250和640并不影響通過線粒體膜極性標志物MTS評估的細胞活力。由于體內PIP作用的瞬時特性,我們關注了可能與降低的細胞活力有關的可能更好地限定早期細胞變化的細胞信號;半胱天冬酶級聯反應的活化是上皮細胞中細胞凋亡事件的早期步驟。我們測量了將肽250或肽640以顯示為降低血糖(圖8)、增加pMLC的程度(圖9)且增強胰島素進入門靜脈的攝取(圖10)的濃度進行頂部暴露60分鐘之后分離的大鼠腸組織中的半胱天冬酶-3的活性水平。在類似的20mM的PIP肽250或640進行頂部暴露之后,腸組織沒有顯示半胱天冬酶-3活性的誘導,而通過ILI施用的對于半胱天冬酶-3的誘導用作正對照的潮霉素(150mg/mL)卻引起了這種酶活性的增加(圖11)。在注射胰島素與20mM的PIP肽250或640、或者單獨的胰島素之后,測量門靜脈中內毒素的濃度。在治療組中沒有觀察到區別。
結論
為了增強生物藥物的口服遞送的目的,已經探索了多種試劑以增加腸細胞旁通量;皂樹皂苷(quilaja saponin)、甘草酸二鉀、18β-甘草(glycyrrhetinic)、癸酸鈉、牛磺酸和烷基麥芽糖苷(alkylmaltosides)僅是已經描述的試劑的一小部分。總的來說,這些試劑通常用體外細胞體系(諸如Caco-2單層或分離的腸組織)進行篩選。這些試劑中的一些,如棕櫚酰肉堿,最初顯示出希望,但最終其優勢與對細胞膜的裂解作用有關并降低了細胞活力。人乳中存在的中鏈脂肪酸癸酸鈉,甚至已經被批準作為直腸氨芐青霉素栓劑中的吸收增強劑。雖然癸酸鹽顯示引起TJ擴張,以增強體外細胞旁透過性,但是在體內癸酸鹽對于其在人中的作用的功效與細胞旁透過性修飾相比,與直腸粘膜的非特異性損傷具有更好的相關性。目前,沒有設計為在口服遞送之后增強生物藥物的細胞旁轉運的試劑被批準。
最初基于其作用機理的不確定性的毒性似乎是限制藥劑鑒定為在口服遞送之后安全地增強細胞旁轉運的核心要素。鑒于此,最近的研究表明癸酸鹽可以改變TJ成分三細胞素(tricellulin)的表達,以通過上皮中的三細胞接觸來增加細胞旁通量。這一發現可能解釋癸酸鹽在體外的優點,因為其與由模仿了TJ蛋白claudin 1的細胞外環結構域的肽誘導的體外和體內的細胞旁攝取可能增加類似,所述肽。因此,選擇性解構三細胞TJ接觸的方法可提供安全且有效地增加細胞旁透過性的潛在作用機理。我們已經采取了這樣的方法以確定具有限定的作用機理的藥劑,其可以瞬時地打開腸TJ以增強生物藥物的細胞旁攝取。我們的方法是基于最初由Papenheimer做出的發現,其說明腸上皮頂部具有內源性營養活化的機理以增加溶質的細胞旁透過。隨后的研究顯示這樣的細胞旁透過的增加是由于肌球蛋白輕鏈(MLC)磷酸化的增加。
在控制TJ-介導的溶質透過性中MLC磷酸化的驗證,最初使用合理設計的、穩定的MLC激酶(PIK)肽的膜透過性抑制劑來確認,該透過性抑制劑在慢性上皮炎癥模型中顯示有效。目前,我們的目標是確認膜透過性的、穩定的MLC磷酸酶(MLCP)的選擇性抑制劑,其抵消殘余的MLC激酶活性的作用(圖12)。我們采用公認的原則來鑒定能夠選擇性調節蛋白質與蛋白質之間相互作用的短肽序列,目的是模擬涉及MLCP調節蛋白(CPI-17和MYPT)的界面接觸。通過這些調節元件的MLCP調節為上皮細胞提供了至少兩個不同的控制MLC磷酸化的機理,從而改變了TJ的功能性特性。具有足以實現靶特異性的廣泛界面接觸位點的肽具有差的膜透過性,并且進一步限制了腸腔中和上皮細胞細胞質中的肽酶-介導的分解代謝。我們設計并測試了模擬內源性序列以增加特異性的肽組,其由D-型氨基酸制備用于增加穩定性,并且包含提高的正/負電荷比以增加膜透過性。
目前的研究已經證實了在活細胞中使用穩定的膜透過性肽以干擾界面接觸位點并改變蛋白質磷酸酶1的方法。如在先所示以設計為靶向RVxF-型PP1-結合基序的膜透過性肽或者其他細胞膜透過性肽的MLPC調節而言,在本研究中檢測的PIP肽在體外或體內沒有顯示任何細胞毒性作用。這與微囊藻毒素形成鮮明對比,微囊藻毒素是一類通過結合常見位點而抑制多種PP1以及其它Ser/Thr蛋白磷酸酶的環狀七肽肝毒素;與微囊藻毒素中毒相關的疾病與由多種蛋白質的磷酸化增加而引起的非特異性作用相關。我們的結果說明了,通過靶向MLCP和不誘導細胞毒性(可能由于它們的特定作用)的特異性調節蛋白之間的界面接觸而選擇性抑制MLCP活性,增強生物藥物的口服攝取的可行性。
在過去20年的調查表明,Ca2+/鈣調蛋白活化MLCK的活性,而Rho激酶和蛋白激酶C分別通過MYPT1和CPI-17調節MLCP。當與肽640的作用相比,肽250具有較小的動態作用并打開TJ結構以轉運更大的溶質。這樣的數據表明,與那些通過CPI-17介導的相比,通過Rho激酶有關的機理的MLCP抑制引起了更為持久和廣泛的TJ功能的修飾(圖12)。這些研究與Rho A可通過其對MYPT1的作用來介導緩慢的、持久的TJ改變的可能性是一致的,而PKC對CPI-17的作用可為JI功能中更迅速且動態的改變提供機理。PIP肽250和640作用中的差別在其中它們的濃度和持續暴露可以被控制的體外研究中更加顯而易見。體內研究與關于PIP肽作用的動態性質的體外觀察結果一致,但不是顯而易見的。這可能是由于體內所經歷條件的增加的可變性。
在這些研究中鑒定和測試的PIP肽已經為支持關于MLC磷酸化的調控的幾種概念提供了主要成果的證明。1)為干擾PP1及其調節蛋白CPI-17以及MYPT1之間的蛋白質-蛋白質相互作用而設計的肽能夠瞬時地增加MLC的磷酸化。2)由這些肽誘發的MLC磷酸化的增加能夠增加大分子的細胞旁通量。3)由這些為干擾PP1及其調節蛋白CPI-17以及MYPT1之間的蛋白質-蛋白質相互作用而設計的肽誘導的細胞旁透過性改變的調控是迅速且可逆的。4)與這些對細胞旁屏障性質的動態效果相關聯的MLC磷酸化中的改變與細胞毒性無關,或者不導致細胞毒性。最后這一點在下一步的考慮中是非常重要的。雖然PIP肽640和250是有效且明顯為特異性和非毒性的,但是對其功能而言所需的濃度在mM的范圍內。這實際上并不是一個重要的問題,因為在其中同時定位生物藥物的局部應用中實現這些濃度是易于實現的。優化這些PIP肽以降低所需的濃度是未來感興趣的一個領域。這樣的效果將需要多重和反復的努力以使肽結構最小化,同時保持靶特異性和作用以及增加膜透過性的效率以達到所需的細胞內靶標。這些努力可能得益于使用穩定的磷酸化氨基酸類似物,而不是使用帶負電的氨基酸來模擬這些所需的功能要素。
總的來說,我們的研究表明,合理設計阻斷CPI-17或MYPT1與PP1(MLCP)的相互作用的肽能夠產生關于TEER和細胞旁轉運的不同的藥理學結果。雖然CPI-17在平滑肌中廣泛表達,但是該數據是第一次表明其在分化的上皮細胞中的功能性作用。此外,我們的結果表明,可能存在獨立的調節機理,其允許CPI-17和MYPT1動態地打開TJ至各種程度和速率。最終,我們已經表明,使用設計為增加MLCP活性的肽操縱MLCP功能可以應用于增加胰島素的口服生物利用度的藥學相關問題。
序列表
<110> 巴斯大學
<120> 藥物遞送增強劑
<130> P021142WO
<160> 21
<170> PatentIn 3.5版
<210> 1
<211> 5
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<220>
<221> MOD_RES
<222> (1)..(1)
<223> 磷酸化
<220>
<221> 變體
<222> (1)..(1)
<223> Xaa1 為任意帶負電的氨基酸殘基, 例如 pTyr,
Asp 或 Glu
<220>
<221> 變體
<222> (2)..(2)
<223> Xaa2 為任意小的疏水性氨基酸殘基, 例如 Val
或 Ala
<220>
<221> 變體
<222> (3)..(3)
<223> Xaa3 為任意帶正電的氨基酸殘基, 例如
Lys 或 Arg
<220>
<221> 變體
<222> (4)..(4)
<223> Xaa4 為任意親水性氨基酸殘基, 例如Tyr, Phe,
Thr 或 Ser
<220>
<221> 變體
<222> (5)..(5)
<223> Xaa4 為任意帶負電的氨基酸殘基, 例如
PTyr, Asp 或 Glu
<400> 1
Tyr Val Lys Tyr Tyr
1 5
<210> 2
<211> 4
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<220>
<221> 變體
<222> (1)..(1)
<223> Xaa 在位置1 為任意氨基酸殘基
<400> 2
Xaa Tyr Gln Tyr
1
<210> 3
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<400> 3
Arg Arg Val Glu Val Lys Tyr Asp Arg Arg
1 5 10
<210> 4
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<400> 4
Arg Lys Ala Lys Tyr Gln Tyr Arg Arg Lys
1 5 10
<210> 5
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<220>
<221> 變體
<222> (1)..(1)
<223> Xaa 為至多10個帶正電的氨基酸殘基
<220>
<221> 變體
<222> (2)..(2)
<223> Xaa2 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (3)..(3)
<223> Xaa3 不存在或為疏水性氨基酸殘基
<220>
<221> 變體
<222> (4)..(4)
<223> Xaa4為帶負電的氨基酸殘基
<220>
<221> 變體
<222> (5)..(5)
<223> Xaa5 為小的疏水性氨基酸殘基
<220>
<221> 變體
<222> (6)..(6)
<223> Xaa6 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (7)..(7)
<223> Xaa7 為疏水性氨基酸殘基
<220>
<221> 變體
<222> (8)..(8)
<223> Xaa8 為帶負電的氨基酸殘基
<220>
<221> 變體
<222> (8)..(8)
<223> Xaa8 為帶負電的氨基酸殘基
<220>
<221> 變體
<222> (9)..(9)
<223> Xaa9 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (10)..(10)
<223> Xaa10 為至多10個帶正電的氨基酸殘基
<400> 5
Lys Lys Ala Asp Ala Lys Ala Asp Lys Lys
1 5 10
<210> 6
<211> 8
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<220>
<221> 變體
<222> (1)..(1)
<223> Xaa1 不存在或為疏水性氨基酸殘基
<220>
<221> 變體
<222> (2)..(2)
<223> Xaa2 為帶負電的氨基酸殘基
<220>
<221> 變體
<222> (3)..(3)
<223> Xaa3 為小的疏水性氨基酸殘基
<220>
<221> 變體
<222> (4)..(4)
<223> Xaa4 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (5)..(5)
<223> Xaa5 為疏水性氨基酸殘基
<220>
<221> 變體
<222> (6)..(6)
<223> Xaa6 為帶負電的氨基酸殘基
<220>
<221> 變體
<222> (7)..(7)
<223> Xaa7 為帶正電的氨基酸殘基
<400> 6
Ala Asp Ala Lys Ala Asp Lys Lys
1 5
<210> 7
<211> 8
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<220>
<221> 變體
<222> (1)..(1)
<223> Xaa1 為至多10個帶正電的氨基酸殘基
<220>
<221> 變體
<222> (2)..(2)
<223> Xaa2 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (3)..(3)
<223> Xaa3 不存在或為疏水性氨基酸殘基
<220>
<221> 變體
<222> (4)..(4)
<223> Xaa4 為帶負電的氨基酸殘基
<220>
<221> 變體
<222> (5)..(5)
<223> Xaa5 為小的疏水性氨基酸殘基
<220>
<221> 變體
<222> (6)..(6)
<223> Xaa6 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (7)..(7)
<223> Xaa7 為疏水性氨基酸殘基
<220>
<221> 變體
<222> (8)..(8)
<223> Xaa8 為帶負電的氨基酸殘基
<400> 7
Lys Lys Ala Asp Ala Lys Ala Asp
1 5
<210> 8
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<400> 8
Arg Val Thr Val Lys Tyr Asp Arg Arg
1 5
<210> 9
<211> 12
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<220>
<221> 變體
<222> (1)..(1)
<223> Xaa1 為至多10個帶正電的氨基酸殘基
<220>
<221> 變體
<222> (2)..(2)
<223> Xaa2 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (3)..(3)
<223> Xaa3 為 Ala 或 Val
<220>
<221> 變體
<222> (4)..(4)
<223> Xaa4 不存在或為 Ala 或 為1、2、3 或 4個帶正電的
氨基酸殘基
<220>
<221> 變體
<222> (6)..(6)
<223> Xaa6 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (6)..(6)
<223> Xaa6 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (9)..(9)
<223> Xaa9 不存在或為 Ala 或 為1、2、3 或 4個帶正電的
氨基酸殘基
<220>
<221> 變體
<222> (10)..(10)
<223> Xaa10 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (12)..(12)
<223> Xaa12 為至多10個帶正電的氨基酸殘基
<400> 9
Lys Lys Ala Ala Lys Tyr Gln Tyr Ala Lys Lys Lys
1 5 10
<210> 10
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<220>
<221> 變體
<222> (1)..(1)
<223> Xaa1 為至多10個帶正電的氨基酸殘基
<220>
<221> 變體
<222> (2)..(2)
<223> Xaa2 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (3)..(3)
<223> Xaa3 為Ala 或 Val
<220>
<221> 變體
<222> (4)..(4)
<223> Xaa4 不存在或 為Ala 或為 1、2、3、或 4個帶正電的
氨基酸殘基
<220>
<221> 變體
<222> (5)..(5)
<223> Xaa5 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (5)..(5)
<223> Xaa5 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (9)..(9)
<223> Xaa9 不存在或為Ala 或為 1、2、3、或 4個帶正電的
氨基酸殘基
<400> 10
Lys Lys Ala Ala Lys Tyr Gln Tyr Ala
1 5
<210> 11
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<220>
<221> 變體
<222> (1)..(1)
<223> Xaa1 為Ala 或 Val
<220>
<221> 變體
<222> (1)..(1)
<223> Xaa1 為Ala 或 Val
<220>
<221> 變體
<222> (2)..(2)
<223> Xaa2 不存在或 為Ala 或 為1、2、3 或 4個帶正電的
氨基酸殘基
<220>
<221> 變體
<222> (3)..(3)
<223> Xaa3 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (7)..(7)
<223> Xaa7 不存在或為 Ala, 或為 1、2、3、或 4個帶正電的
氨基酸殘基
<220>
<221> 變體
<222> (8)..(8)
<223> Xaa8 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (8)..(8)
<223> Xaa8 為帶正電的氨基酸殘基
<220>
<221> 變體
<222> (10)..(10)
<223> Xaa10 為至多10個帶正電的氨基酸殘基
<400> 11
Ala Ala Lys Tyr Gln Tyr Ala Lys Lys Lys
1 5 10
<210> 12
<400> 12
000
<210> 13
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<400> 13
Arg Val Thr Val Lys Tyr Asp Arg Arg
1 5
<210> 14
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<400> 14
Arg Arg Val Thr Val Lys Tyr Asp Arg Arg
1 5 10
<210> 15
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<220>
<221> MOD_RES
<222> (4)..(4)
<223> 磷酸化
<400> 15
Arg Arg Val Thr Val Lys Tyr Asp Arg Arg
1 5 10
<210> 16
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<400> 16
Arg Arg Val Thr Val Lys Tyr Lys Arg Arg
1 5 10
<210> 17
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<220>
<221> MOD_RES
<222> (4)..(4)
<223> 磷酸化
<400> 17
Arg Arg Val Thr Val Lys Tyr Lys Arg Arg
1 5 10
<210> 18
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<400> 18
Arg Arg Lys Thr Val Lys Tyr Asp Arg Arg
1 5 10
<210> 19
<211> 6
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<400> 19
Lys Ala Lys Tyr Gln Tyr
1 5
<210> 20
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<400> 20
Arg Arg Asp Tyr Lys Val Glu Val Arg Arg
1 5 10
<210> 21
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 藥物遞送增強肽
<400> 21
Lys Arg Arg Tyr Gln Tyr Lys Ala Lys Arg
1 5 10
- 還沒有人留言評論。精彩留言會獲得點贊!