頭孢菌素化合物的合成的制作方法
本申請要求2014年8月15日提交的美國臨時申請62/037,676、2014年10月20日提交的美國臨時申請62/065,993和2015年2月4日提交的美國臨時申請62/111,840的權益,其內容通過引用以其整體并入本申請。2.
技術領域:
本公開涉及經由鈀催化的偶聯反應的頭孢菌素的合成。3.
背景技術:
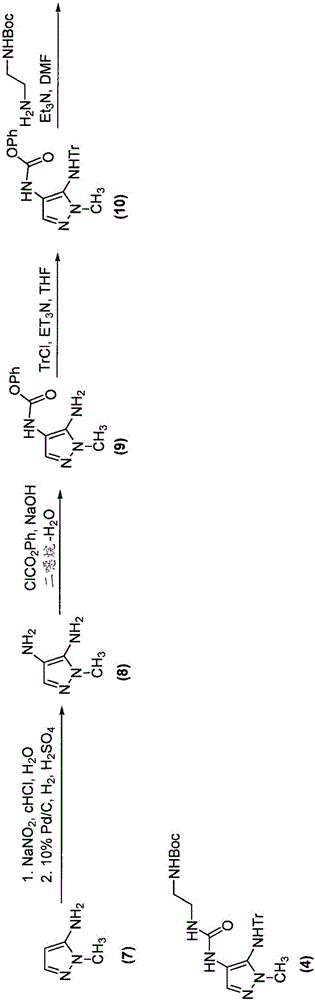
:含有式(I)的化學基礎結構的頭孢菌素化合物是重要的抗菌治療劑。若干種已知的頭孢菌素化合物的制備涉及由在如下結構中的C1所指示的烯丙基碳處形成新的鍵:該烯丙基部分存在于例如頭孢洛扎(ceftolozane)中,所述頭孢洛扎是一種頭孢菌素抗菌劑,也被稱為CXA-101、FR264205或化學名稱諸如(6R,7R)-3-[(5-氨基-4-{[(2-氨基乙基)氨基甲酰基]氨基}-1-甲基-1H-吡唑-2-鎓-2-基)甲基]-7-({(2Z)-2-(5-氨基-1,2,4-噻二唑-3-基)-2-[(1-羧基-1-甲基乙氧基)亞氨基]乙酰基}氨基)-8-氧代-5-硫雜-1-氮雜二環[4.2.0]辛-2-烯-2-羧酸鹽和7β-[(Z)-2-(5-氨基-1,2,4-噻二唑-3-基)-2-(1-羧基-1-甲基乙氧基亞氨基)乙酰氨基]-3-{3-氨基-4-[3-(2-氨基乙基)脲基]-2-甲基-1-吡唑鎓基}甲基-3-頭孢烯-4-羧酸鹽。頭孢洛扎硫酸鹽是化合物(VII)的藥學上可接受的頭孢洛扎鹽,其可經配制以用于靜脈內給藥或輸注。頭孢洛扎可以使用美國專利7,129,232和7,192,943以及Toda等人,“SynthesisandSARofnovelparenteralanti-pseudomonalcephalosporin’s:DiscoveryofFR264205,”Bioorganic&MedicinalChemistryLetters,18,4849-4852(2008)中描述的方法獲得,其各自通過引用以其整體并入本申請。這些方法例示于圖1A和1B中。仍然需要發現新的用于合成包含式(I)的化學基礎結構的頭孢菌素化合物(例如,頭孢洛扎)的制備方法。4.技術實現要素:本申請提供了采用鈀-催化的烷基化反應以合成式(I)的頭孢菌素化合物的方法以及與其相關的組合物。在一個方面中,本申請提供了一種用于制備式(II)的化合物或其鹽的方法,包括如下步驟:在包含(a)鈀源和(b)結合鈀的配體的試劑的存在下使式(III)的化合物或其鹽與親核試劑(Nuc)混合,例如使式(III)的化合物或其鹽與親核試劑(Nuc)反應:以形成式(II)的化合物或其鹽。在另一個方面中,本申請提供了一種組合物,其包含式(Va)的化合物和鈀。在另一個方面中,本申請提供了一種用于制備式(VIII)的化合物或其鹽的方法:包括如下步驟:在包含(a)鈀源和(b)結合鈀的配體的試劑的存在下使式(IX)的化合物或其鹽與親核試劑(Nuc)(例如R4–M)混合,例如使式(IX)的化合物或其鹽與親核試劑(Nuc)(例如R4–M)反應:以形成式(VIII)的化合物或其鹽。所述親核試劑、鈀源、結合鈀的配體以及式(II)、(III)、(VIII)和(IX)的化合物的變量在本申請中定義。在一些實施方案中,所述方法包括鈀的去除。在一些實施方案中,所述方法包括鈀的回收。5.附圖說明圖1A是一個示例性合成方案,其展示頭孢洛扎合成的已知方法(參見,例如,美國專利7,129,232和7,192,943以及Toda等人,"SynthesisandSARofnovelparenteralanti-pseudomonalcephalosporins:DiscoveryofFR264205,"Bioorganic&MedicinalChemistryLetters,18,4849-4852(2008))。圖1B是用于制備頭孢洛扎起始物質,即一種經保護的5-氨基-1-甲基吡唑的合成方案。(參見,例如,Toda等人,"SynthesisandSARofnovelparenteralanti-pseudomonalcephalosporins:DiscoveryofFR264205,"Bioorganic&MedicinalChemistryLetters,18,4849-4852(2008))。圖2是展示TATD-CLE和TATD-TFA的消耗的HPLC跡線圖。圖3是TATD-QUATE的最終的有機層的HPLC跡線圖。圖4是展示叔丁基頭孢洛扎TFA的消耗的HPLC跡線圖。圖5是展示頭孢洛扎TFA的干燥濾餅的純度的HPLC跡線圖。圖6是分離的頭孢洛扎TFA的參比1HNMR譜圖。圖7是化合物3taz的HPLC色譜圖。顯示在9.552min的主峰。圖8是在15分鐘反應時間后,由化合物3taz形成化合物5taz的反應混合物的HPLC色譜圖。顯示在1.777min的主峰。6.具體實施方式本申請提供了使用鈀催化取代式(I)的化合物的C1位的方法以及相關的組合物。本申請提供的方法提供了優于先前公開的方法的若干優點,包括相比于例如如圖1A中所示的用于將化合物3轉化為化合物5a的類似反應而言更高的產率、更高的純度、更快的反應時間以及使用更少量的固體試劑。在一個方面中,本申請提供了一種用于制備式(II)的化合物或其鹽的方法:包括如下步驟:在包含(a)鈀源和(b)結合鈀的配體的試劑的存在下使式(III)的化合物或其鹽與親核試劑(Nuc)(例如R4–M)混合,例如使式(III)的化合物或其鹽與親核試劑(Nuc)(例如R4–M)反應:以形成式(II)的化合物或其鹽。所述親核試劑、鈀源、結合鈀的配體以及式(II)和(III)的化合物的變量在下文中定義。親核試劑(Nuc)親核試劑包括羧酸、醇、水、酰胺、脲、硫醇、含N的雜芳基化合物(例如任選取代的吡唑)和含N的雜環化合物。這些親核試劑可以以陰離子形式使用,例如作為羧酸鹽、氫氧化物和醇鹽。在一個實施方案中,Nuc是R4-M,其中:M是H、金屬陽離子、非金屬陽離子或孤對電子(lonepairofelectrons);且R4選自羧酸根、氫氧根、烷醇、脲、含N的雜芳基和含N的雜環基,其中所述羧酸根、烷醇、硫醇、含N的雜芳基和含N的雜環基是任選取代的。M可以是選自例如堿金屬、堿土金屬、過渡金屬和主族金屬的金屬。對于具有大于1(例如,2)的有效電荷的金屬陽離子而言,多于一當量的R4將會存在于Nuc(例如(R4)2M)中。本領域技術人員將會認識到當孤對反應形成鍵時,R4的有效電荷會發生變化。在一個實施方案中,M是H、金屬陽離子或非金屬陽離子并且R4選自:在另一個實施方案中,M是H并且R4-M選自和H2O。在另一個實施方案中,M是孤對電子并且R4-M是:在另一個實施方案中,M是孤對電子并且R4-M是式(X)的化合物:其中R5是氮保護基;和R6是氮保護基。在一些實施方案中,式(X)的化合物具有式(UBT)的結構:該親核試劑,即一種經保護的4-氨基-5-氨基-1-甲基吡唑的合成的一個實例示于圖1B中。變量LGLG是離去基團。在某些實施方案中,LG是鹵素、-O(C=O)N(R18)2、-O(C=O)OR18和-OC(O)R18,其中R18在每種情況下獨立地選自C1-6烷基和鹵代烷基(例如-CF3)。在某些實施方案中,LG是鹵素或-OC(O)R18,其中R18選自C1-6烷基和鹵代烷基(例如-CF3)。在一個具體實施方案中,LG是氯或–OC(O)CF3。在一些實施方案中,鹵代烷基是鹵代C1-6烷基。變量R1在某些實施方案中,R1是R1’-Z;其中R1’選自鍵、芳基、環烷基、環烯基、雜環基和雜芳基;其中Z是1-2個取代基,其每次出現時獨立地選自氫、鹵素、羥基、羥基烷基、氨基烷基、烷基、亞烷基、烯基、雜烷基、氰基和氨基,其中Z任選獨立地被氨基、鹵素、羧基、羧酰胺基團、氧代、氮保護基、氧保護基或-P(O)(OZ’)2取代一次或多次;和其中Z’獨立地是氫或氧保護基。在一個實施方案中,R1選自芳基和雜芳基。在某些實施方案中,R1是被取代的或未被取代的選自下面的芳基或雜芳基:噻吩基、呋喃基、噻唑基、四唑基、噻二唑基、吡啶基、苯基、苯酚基團、環己二烯基和二硫雜環丁基。在一個具體實施方案中,R1選自下述基團:及其鹽,其中Z’如本申請中所定義。在一些實施方案中,R1選自:在一些實施方案中,R1是在一個具體實施方案中,R1是在另一個具體實施方案中,R1是在一些實施方案中,Z’是C1-6烷基,諸如Me、Et或叔丁基。在一些實施方案中,Z’是對堿不穩定的氧保護基,諸如但不限于-COMe、-COEt和-(CO)-丙基。在一些實施方案中,Z’是在還原條件(例如催化氫化)下去除的氧保護基,諸如芐基。在一些實施方案中,Z’是對酸不穩定的氧保護基,諸如但不限于叔丁基、4-甲氧基芐基、2-甲氧基芐基或三苯基甲基,優選叔丁基。變量R2在某些實施方案中,R2選自氫和烷氧基。在該方法的一個具體實施方案中,R2是氫。變量Y在某些實施方案中,Y選自鍵、CH2、CH2S、SCH2、C=C(H)CH2CO2R’、CH(OR’)、C=N(OR’)、CHN(R”)2和C=NR”;其中R’選自氫、氧保護基和烷基,其中所述烷基被鹵素、羥基或-CO2R15任選取代一次或多次;其中R”是取代基,其每次出現時選自氫、烷基、C(O)雜環基和氮保護基,其中任意兩個R”取代基可以組合形成環或單個氮保護基;和其中R15獨立地是氫或氧保護基。在一些實施方案中,R’是C1-6烷基,諸如Me、Et或叔丁基。在一些實施方案中,R’是對堿不穩定的氧保護基,諸如但不限于-COMe、-COEt和-(CO)-丙基。在一些實施方案中,R’是在還原條件(例如,催化氫化)下去除的氧保護基,諸如芐基。在一些實施方案中,R’是對酸不穩定的氧保護基,諸如但不限于叔丁基、4-甲氧基芐基、2-甲氧基芐基或三苯基甲基,優選叔丁基。在一些實施方案中,Y選自:-CH2-、在所述方法的一個實施方案中,Y選自鍵、CH2、CH2S和C=C(H)CH2CO2R’,并且R’選自氫和氧保護基。在另一個實施方案中,Y是C=N(OR’)并且R’選自氧保護基、氫、甲基、乙基、CH2CO2R15和C(CH3)2CO2R15。在另一個實施方案中,Y是CH(OR’)并且R’是氫或氧保護基。在另一個實施方案中,Y是CHN(R”)2并且R”在每次出現時獨立地選自氫、氮保護基和在一個具體實施方案中,Y是C=N(OR’)并且R’是C(CH3)2CO2tBu。在一些實施方案中,Y是變量R3在某些實施方案中,R3選自氫和氧保護基。在一些實施方案中,R3是C1-6烷基,諸如Me、Et或叔丁基。在一些實施方案中,R3是對堿不穩定的氧保護基(即,在堿性條件下去除的保護基),諸如但不限于Me、Et和丙基。在一些實施方案中,R3是可以在氫化條件下去除的保護基,諸如芐基。在一些實施方案中,R3是對酸不穩定的氧保護基(即,在酸性條件下去除的保護基),諸如但不限于叔丁基二甲基甲硅烷基、叔丁基、4-甲氧基芐基、2-甲氧基芐基或三苯基甲基。在該方法的另一個實施方案中,R3是選自芐基醚的氧保護基。芐基醚可以是被取代的(例如,被一個或多個烷氧基取代基取代)或未被取代的。在一個具體實施方案中,R3是(即,4-甲氧基芐基、PMB、MPM)。變量R4R4是如下面所述的由Nuc(例如R4–M)的添加所產生的基團。在一個實施方案中,R4是羧基、羥基、烷氧基、脲、脲加合物、含N的雜芳基或含N的雜環基,其中所述羧基、烷氧基、含N的雜芳基和含N的雜環基是任選取代的。在一些實施方案中,R4是含N的雜芳基(即,含氮的雜芳基或在環中含有至少一個氮的雜芳基)。含氮的雜芳基化合物包括但不限于吡唑類、吡咯類、三唑類、吡啶類、嘧啶類、噻唑類和噻二唑類,其各自可以是任選取代的。在一些實施方案中,R4是吡唑基、吡咯基、三唑基或吡啶基,其各自是任選取代的。在一些實施方案中,R4是吡唑基或吡啶基,其各自是任選取代的。在一些實施方案中,R4是未被取代的吡啶基。在一些實施方案中,含N的雜芳基通過雜芳基環N原子連接至式(II)的化合物的其余部分(即,環N-連接的含氮的雜芳基)。在一個實施方案中,R4選自其中R5是氮保護基;和R6是氮保護基。在一個實施方案中,R4是:在一些實施方案中,R4是被取代的吡唑基。在一些實施方案中,R4是具有一個、兩個、三個或四個取代基的被取代的吡唑基。在一些實施方案中,R4是被C1-6烷基取代的吡唑基。在一些實施方案中,R4是被脲取代的吡唑基。在一些實施方案中,R4是被胺取代的吡唑基。在一個實施方案中,R4是其是由Nuc(即,R4-M,其中M是孤對電子)的添加所產生,例如,式(X)的化合物:在一些實施方案中,R5是對酸不穩定的氮保護基。在一些實施方案中,R5是叔丁基氧基羰基。在一些實施方案中,R6是對酸不穩定的氮保護基。在一些實施方案中,R6是三苯基甲基。在一些實施方案中,R5和R6各自獨立地是對酸不穩定的氮保護基。在一些實施方案中,R5和R6各自獨立地是三苯基甲基、叔丁基、叔丁氧基羰基、2-三甲基甲硅烷基乙氧基羰基或4-甲氧基芐基氧基羰基。在一個實施方案中,R5是叔丁基氧基羰基并且R6是三苯基甲基。在一個實施方案中,R’是叔丁基;R3是4-甲氧基芐基醚(即,4-甲氧基芐基);R5是叔丁基氧基羰基;和R6是三苯基甲基。在一些實施方案中,式(II)的化合物具有式(II’)的結構:其中R1、R2、R3、R4和R’如本申請中所述。鈀源術語“鈀源”表示鈀的來源。在一些實施方案中,所述鈀源是已知會促進鈀π-烯丙基化學的任何來源,包括但不限于可溶于溶劑中的鈀催化劑和在固體載體上的鈀,諸如鈀(0)/炭黑粉。可溶于溶劑中的鈀催化劑包括鈀(II)催化劑,諸如二(乙腈)二氯化鈀(II)、二(乙酰基丙酮酸)鈀(II)、二(芐腈)二氯化鈀(II)、二(二亞芐基丙酮)鈀、乙酸鈀(II)、三氟乙酸鈀(II)、烯丙基二氯化鈀(II)二聚體、二氯化鈀(II)、二溴化鈀(II)、四(乙腈)四氟硼酸鈀(II)、[1,2-二(二苯基膦基)乙烷]二氯化鈀(II)、1,1’-二(二苯基膦基)二茂鐵-二氯化鈀(II)二氯甲烷加合物、二(三環己基膦)鈀(0)、二(三乙基膦)二氯化鈀(II)、二(三苯基膦)乙酸鈀(II)、二(三苯基膦)二氯化鈀(II)、二[三(鄰-甲苯基)膦]二氯化鈀(II)、二氯二(三環己基膦)鈀(II)和反式-芐基(氯)二(三苯基膦)鈀(II),以及鈀(0)催化劑,諸如四(三苯基膦)鈀(0)、二[1,2-二(二苯基膦基)乙烷]鈀(0)、二(三-叔丁基膦)鈀(0)、二(二亞芐基丙酮)鈀(0)和三(二亞芐基丙酮)二鈀(0)(即,Pd2dba3,呈游離和溶劑化物形式,例如,作為氯仿加合物)。例如,所述鈀源可以包含三(二亞芐基丙酮)二鈀(0),例如,由三(二亞芐基丙酮)二鈀(0)組成或基本上由三(二亞芐基丙酮)二鈀(0)組成。在一個實施方案中,所述鈀源包含鈀(0)或鈀(II)鹽,任選作為絡合物(例如,進一步包含配體)。鈀源的非限定性列舉包括:烯丙基二氯化鈀(II)二聚體、二(乙腈)二氯化鈀(II)、二(乙酰基丙酮酸)鈀(II)、二(芐腈)二氯化鈀(II)、二(二亞芐基丙酮)鈀、乙酸鈀(II)、三氟乙酸鈀(II)、二氯化鈀(II)、二溴化鈀(II)、四(乙腈)四氟硼酸鈀(II)、三(二亞芐基丙酮)二鈀(0)、三(二亞芐基丙酮)二鈀(0)-氯仿加合物、[1,2-二(二苯基膦基)乙烷]二氯化鈀(II)、1,1’-二(二苯基膦基)二茂鐵-二氯化鈀(II)二氯甲烷加合物、二(三環己基膦)鈀(0)、二(三乙基膦)二氯化鈀(II)、二(三苯基膦)乙酸鈀(II)、二(三苯基膦)二氯化鈀(II)、二(三-叔丁基膦)鈀(0)、二[1,2-二(二苯基膦基)乙烷]鈀(0)、二[三(鄰-甲苯基)膦]二氯化鈀(II)、二氯二(三環己基膦)鈀(II)、四(三苯基膦)鈀(0)、四(三乙基膦)鈀(0)和反式-芐基(氯)二(三苯基膦)鈀(II)。在一個實施方案中,所述鈀源選自烯丙基二氯化鈀(II)二聚體、三(二亞芐基丙酮)二鈀(0)、三(二亞芐基丙酮)二鈀(0)-氯仿加合物、四(三苯基膦)鈀、乙酸鈀(II)、三氟乙酸鈀(II)、二氯化鈀(II)、二溴化鈀(II)和二(乙腈)二氯化鈀(II)。在一個實施方案中,所述鈀源是三(二亞芐基丙酮)二鈀(0)。在一些實施方案中,鈀-催化的反應提供了金屬介導的反應,其與非金屬催化的反應相比,提供更高的產率和/或期望產物的更潔凈的形成。通常,鈀-催化的反應以亞化學計量的量的鈀源和一種或多種結合鈀的配體進行以促進催化循環。在一些實施方案中,鈀-催化促進兩種試劑的反應以提供更高分子量的產物。在一些實施方案中,兩種試劑的量是相同的。在一些實施方案中,兩種試劑的量是不同的。在兩種試劑的量不同的情況下,量更少的試劑被稱為限制性試劑。在一些實施方案中,所述鈀源以相對于式(III)的化合物而言約0.2摩爾%至約5摩爾%的量存在。在一些實施方案中,所述鈀源以相對于式(III)的化合物而言約0.2摩爾%至約1.5摩爾%的量存在。在一個實施方案中,所述鈀源以相對于式(III)的化合物而言約0.5摩爾%至約5摩爾%的量存在。在另一個實施方案中,所述鈀源以相對于式(III)的化合物而言約0.5摩爾%至約1.5摩爾%的量存在。在另一個實施方案中,所述鈀源以相對于式(III)的化合物而言約1.0摩爾%的量存在。在一些實施方案中,所述鈀源與限制性試劑的摩爾量相比以約0.2%、約0.5%、約1%、約2%、約3%、約4%或約5%鈀的摩爾量存在。在一個示例性實施例中,2mol%四(三苯基膦)鈀(0)是0.02摩爾的鈀(0)/摩爾的限制性試劑(例如,式(III)的化合物)。在另一個實施例中,1mol%三(二亞芐基丙酮)二鈀是0.02摩爾的鈀(0)/摩爾的限制性試劑。結合鈀的配體結合鈀的配體的作用是穩定鈀催化循環中的中間體物質,同時促進期望的反應產物的形成。配體與金屬的摩爾比可以被修改以最優化反應條件,諸如速率或產率。在一些實施方案中,結合鈀的配體與鈀的摩爾比在約1:1至約10:1,諸如約1.5:1至約5:1、約1.5:1至約4:1、約2:1至約4:1或約3:1至約4:1的范圍內。在一些實施方案中,所述結合鈀的配體是基于砷的配體,諸如三苯胂。可以用于公開的偶聯中的配體是亞磷酸酯類或膦類。合適的膦配體包括三苯基膦、三-叔丁基膦、2-二環己基膦基-2’,6’-二異丙氧基聯苯(RuPhos)、2-二環己基膦基-2’,4’,6’-三異丙基聯苯(XPhos)、4,5-二(二苯基膦基)-9,9’-二甲基黃嘌呤(Xantphos)、1,2-二(二苯基膦基)乙烷(dppe)和1,3-二(二苯基膦基)丙烷(dppp)。在一個優選的實施方案中,所述配體是亞磷酸酯配體。所述結合鈀的配體可以是式(VI)的亞磷酸酯配體:其中R7在每次出現時獨立地選自苯基、雜芳基、雜環基和C1-6烷基,其中所述苯基、雜芳基、雜環基和C1-6烷基任選被鹵素、C1-6烷基、C1-6烷氧基或N(R9)2中的一個或多個取代,并且其中所述苯基和雜芳基任選被稠合的C3-6環烷基或C3-6雜環基進一步取代;R8選自苯基、雜芳基、雜環基和C1-6烷基,其中所述苯基、雜芳基、雜環基和C1-6烷基任選被鹵素、C1-6烷基、C1-6烷氧基或N(R9)2中的一個或多個取代,并且其中所述苯基和雜芳基任選被稠合的C3-6環烷基或C3-6雜環基取代,或R8是其中L選自–(CH2)n–、R8任選通過鍵或-(CH2)n-與一個R7連接以形成一個環,或與每個R7連接以形成兩個環;每個R9是C1-6烷基,或兩個R9可以組合形成3-10元雜環基,其中雜環基包含1-3個氮原子并且任選被C1-6烷基或C(O)-(C1-6烷基)取代;和n是1、2或3。在另一個實施方案中,R7在每次出現時獨立地選自苯基和C1-6烷基,其中所述苯基和C1-6烷基任選被鹵素、C1-6烷基、C1-6烷氧基或N(R9)2中的一個或多個取代并且其中所述苯基任選被稠合的環烷基進一步取代;和R8選自苯基和C1-6烷基,其中所述苯基和C1-6烷基任選被鹵素、C1-6烷基、C1-6烷氧基或N(R9)2中的一個或多個取代,并且其中所述苯基任選被稠合的環烷基取代,或R8是其中L選自–(CH2)n–、R8任選通過鍵或-(CH2)n-與一個R7連接以形成一個環,或與每個R7連接以形成兩個環;每個R9是C1-6烷基,或兩個R9可以組合形成3-10元雜環基,其中雜環基包含1-3個氮原子并且任選被C1-6烷基或C(O)-(C1-6烷基)取代。在另一個實施方案中,R7在每次出現時獨立地為任選被鹵素、C1-6烷基、C1-6烷氧基或N(R9)2中的一個或多個取代的苯基,并且其中所述苯基任選被稠合的環烷基進一步取代;和R8是任選被鹵素、C1-6烷基、C1-6烷氧基或N(R9)2中的一個或多個取代的苯基,并且其中所述苯基任選被稠合的環烷基取代,或R8是其中L選自–(CH2)n–、R8任選通過鍵或-(CH2)n-與一個R7連接以形成一個環,或與每個R7連接以形成兩個環;每個R9是C1-6烷基,或兩個R9可以組合形成3-10元雜環基,其中雜環基包含1-3個氮原子并且任選被C1-6烷基或C(O)-(C1-6烷基)取代。在另一個實施方案中,所述結合鈀的配體是亞磷酸酯配體,其選自:在一些實施方案中,所述結合鈀的配體選自:在一個具體實施方案中,所述結合鈀的配體是在一個實施方案中,所述結合鈀的配體以相對于鈀源而言約3:1至約10:1的摩爾比存在。在另一個實施方案中,所述結合鈀的配體以相對于鈀源而言約3:1、約4:1、約5:1、約6:1、約7:1、約8:1、約9:1或約10:1的摩爾比存在。在一些優選的實施方案中,所述結合鈀的配體以相對于鈀源中的鈀的摩爾量而言約3:1至約6:1的摩爾比存在。在一些優選的實施方案中,所述結合鈀的配體以相對于鈀源中的鈀的摩爾量而言約3:1、約4:1、約5:1或約6:1的摩爾比存在。在一些實施方案中,所述鈀源和所述結合鈀的配體被包括在作為預絡合的鈀催化劑的單一試劑中。在一個示例性實施例中,四(三苯基膦)鈀(0)包括鈀(0)源和結合鈀的配體三苯基膦。在一些實施方案中,所述鈀源和結合鈀的配體包含被加入至混合物中的兩種試劑。在這種情況下,所述鈀源和結合鈀的配體在混合物內形成活性鈀催化劑。在一個優選的實施方案中,所述鈀源包含三(二亞芐基丙酮)二鈀(0)并且結合鈀的配體是鹽添加劑本申請中公開的方法的試劑也可以包含鹽添加劑。所述鹽添加劑可以是鉀鹽、鈉鹽、鋰鹽、銀鹽或銅鹽。合適的鹽包括但不限于三氟乙酸鉀、三氟乙酸鈉、三氟乙酸鋰、三氟甲磺酸鉀、三氟甲磺酸鈉、三氟甲磺酸鋰、三氟甲磺酸銀和硫酸銅。在一個實施方案中,所述鹽選自三氟乙酸鉀、三氟乙酸鈉、三氟乙酸鋰、三氟甲磺酸鉀、三氟甲磺酸鈉、三氟甲磺酸鋰、三氟甲磺酸銀和硫酸銅。在一個具體實施方案中,所述鹽是三氟乙酸鉀。在一個實施方案中,所述鹽添加劑以相對于式(III)的化合物而言約1:1至約5:1的摩爾比存在。在另一個實施方案中,所述鹽添加劑以相對于式(III)的化合物而言約1:1、約2:1、約3:1、約4:1或約5:1的摩爾比存在。在一個具體實施方案中,所述鹽添加劑以相對于式(III)的化合物而言約2:1至約3:1的摩爾比存在。陰離子在每次出現時獨立地是藥學上可接受的陰離子。在一些實施方案中,是氯離子、溴離子、碘離子、硫酸根、硫酸氫根、甲苯磺酸根(即,甲苯磺酸根)、甲磺酸根(即,甲烷磺酸根)、乙二磺酸根、馬來酸根、磷酸根(例如,單磷酸根、磷酸氫根)、酮戊二酸根、三氟乙酸根或三氟甲磺酸根(即,三氟甲烷磺酸根)。在某些實施方案中,選自氯離子、乙酸根、三氟乙酸根和硫酸氫根(即,氫硫酸根)。在一個具體實施方案中,是三氟乙酸根或硫酸氫根(即,HSO4-)。在某些實施方案中,是三氟乙酸根。在某些實施方案中,是硫酸氫根。6.1定義術語"Cx-y烷基"是指未被取代的飽和的烴基,包括直鏈烷基和支鏈烷基,其在鏈中含有x至y個碳。例如,C1-6烷基是具有1-6個碳的烷基。術語"烷氧基"是指具有與其相連的氧的烷基。代表性的烷氧基包括甲氧基、乙氧基、丙氧基、叔丁氧基等。"醚"是通過氧共價連接的兩個烴。因此,使烷基變成醚的烷基的取代基是烷氧基或類似于烷氧基。術語"C1-6烷氧基"是指具有1-6個碳的烷氧基。本申請中使用的術語"鹵代"或"鹵素"是指F、Cl、Br或I。術語"鹵代烷基"是指具有一個或多個,例如一個、兩個或三個鹵素的烷基。"鹵代C1-6烷基"是鹵代烷基,其具有含1-6個碳的烷基。鹵代C1-6烷基包括氯乙基(ClCH2CH2)、氟丙基(FCH2CH2CH2)和三氟甲基(CF3)。術語"亞烷基"是指基團=CRaRb,其中Ra和Rb各自獨立地是氫或烷基。術語"烯基"和"炔基"是指未被取代的不飽和的脂肪族基團,其在長度上類似于上面所述的烷基,但分別含有至少一個雙鍵或三鍵。術語"胺"和"氨基"是領域公知的并且是指未被取代的和被取代的胺及其鹽,例如,可以被如下通式代表的基團:其中Rc、Rd和Re各自獨立地表示氫、烷基、烯基、-(CH2)m-Rf或Rc和Rd與它們所連接的N原子一起構成在環結構中具有4-8個原子的雜環;Rf表示芳基、環烷基、環烯基、雜環基或多環基;和m是0或1-8的整數。在優選的實施方案中,Rc和Rd中只有一個是羰基,例如Rc、Rd和氮一起不形成酰亞胺。在甚至更優選的實施方案中,Rc和Rd(和任選的Re)各自獨立地表示氫、烷基、烯基或-(CH2)m-Rf。在某些實施方案中,所述氨基是堿性的,即具有pKa>7.00的質子化的形式。術語"酰胺基團"、"酰氨基"和"羧酰胺基團"是領域公知的作為氨基-取代的羰基并且包括可以被如下通式代表的基團:其中Rc和Rd如上面所定義。酰胺基團的優選實施方案將不包括可能是不穩定的酰亞胺類。本申請中使用的術語"雜原子"是指除了碳或氫以外的任何元素的原子。優選的雜原子是氮、氧、磷和硫。術語"羥基烷基"是指具有一個或多個,例如一個、兩個或三個羥基(即,-OH)取代基的烷基。術語"氨基烷基"是指具有一個或多個,例如一個、兩個或三個氨基取代基的烷基。術語"羧基"是領域公知的并且是指–COOH。術語"羧基烷基"是指由烷基終止的羧基取代基(即,烷基酯)。本申請中使用的術語"脲"包括可以由如下通式代表的基團:其中Rc、Rd和Re如上面所定義。本申請中使用的術語"芳基"包括5-、6-和7-元被取代的或未被取代的單環芳基,其中該環的每個原子是碳。術語"芳基"也包括具有兩個或更多個環的多環系統,其中兩個或更多個碳是兩個相鄰的環共有的,其中至少一個所述環是芳族的,例如,其它環可以是環烷基、環烯基、環炔基、芳基、雜芳基和/或雜環基。芳基包括苯基、萘基、菲基、苯酚基團、苯胺基團等。本申請中使用的術語"碳環"、"環烷基"、"碳環基"是指非芳族的被取代的或未被取代的環,其中該環的每個原子是碳。術語"C3-6環烷基"是指環中具有3-6個碳的環烷基。示例性的實施例包括環丙基(C3環烷基)和環戊基(C5環烷基)。術語"雜芳基"包括被取代的或未被取代的芳族5-至7-元環結構,更優選5-至6-元環,其環結構包括1-4個雜原子。術語"雜芳基"也包括具有兩個或更多個環的多環系統,其中兩個或更多個碳是兩個相鄰的環共有的,其中至少一個所述環是雜芳族的,例如其它環可以是環烷基、環烯基、環炔基、芳基、雜芳基和/或雜環基。雜芳基包括例如,吡咯基、呋喃基、噻吩基、咪唑基、噁唑基、噻唑基、三唑基、吡唑基、吡啶基、吡嗪基、噠嗪基和嘧啶基等。藥學上可接受的鹽是本領域技術人員已知的。在一些本申請所述的實施方案中,式(II)和(III)的化合物是三氟乙酸鹽。如本申請所用,“保護基”是在一個或多個反應過程中掩蔽官能團的化學反應性的基團。在一個示例性實例中,氮保護基諸如叔丁氧基羰基(即,叔丁基氧基羰基、Boc或BOC)可以在一個步驟中引入以掩蔽一個反應過程中被保護的氮的化學反應性,然后在酸性條件下去除以允許之前被保護的氮進行反應,例如烷基化。保護基可以是本領域中已知的任一種,諸如描述在Wuts,P.G.M.;Greene,T.W.Greene’sProtectiveGroupsinOrganicSynthesis,第四版;JohnWiley&Sons:Hoboken,NewJersey,2007中的那些。氧和氮保護基是本領域技術人員已知的。氧保護基包括但不限于例如甲基醚、被取代的甲基醚(例如,MOM(甲氧基甲基醚)、MTM(甲基硫基甲基醚)、BOM(芐基氧基甲基醚)、PMBM或MPM(對-甲氧基芐基氧基甲基醚)等)、被取代的乙基醚、被取代的芐基醚、甲硅烷基醚(例如,TMS(三甲基甲硅烷基醚)、TES(三乙基甲硅烷基醚)、TIPS(三異丙基甲硅烷基醚)、TBDMS(叔丁基二甲基甲硅烷基醚)、三芐基甲硅烷基醚、TBDPS(叔丁基二苯基甲硅烷基醚)等)、酯(例如,甲酸酯、乙酸酯、苯甲酸酯(Bz)、三氟乙酸酯、二氯乙酸酯等)、碳酸酯、環狀縮醛和縮酮。氮保護基包括但不限于氨基甲酸酯(包括氨基甲酸甲酯、氨基甲酸乙酯和被取代的氨基甲酸乙酯(例如,Troc)等)、酰胺基團、環狀酰亞胺衍生物、N-烷基和N-芳基胺、芐基胺、被取代的芐基胺、三苯甲基胺、亞胺衍生物和烯胺衍生物。在一些實施方案中,所述氧保護基是對堿不穩定的保護基(即,可以在堿性條件下去除的保護基),諸如當用作酯以保護羧酸時的甲基。在一些實施方案中,所述氧保護基是對酸不穩定的氧保護基(即,可以在酸性條件下去除的保護基),諸如叔丁基、4-甲氧基芐基或三苯基甲基。在一些實施方案中,所述氧保護基是氧化還原敏感性氧保護基,諸如在催化氫化條件下去除的芐基醚。在一些實施方案中,所述氧保護基是甲硅烷基醚,諸如用親核的氟化物去除的TBDMS、TIPS或TES。在一些實施方案中,所述氮保護基是對堿不穩定的氮保護基(即,在堿性條件下去除的保護基),諸如氨基甲酸9-芴基甲酯(Fmoc)。在一些實施方案中,所述氮保護基是對酸不穩定的氮保護基(即,在酸性條件下去除的保護基),諸如三苯基甲基、叔丁基、叔丁氧基羰基、2-三甲基甲硅烷基乙氧基羰基(Teoc)或4-甲氧基芐基氧基羰基。在一些實施方案中,所述氮保護基是氧化還原敏感性氮保護基,諸如可以在催化氫化條件下去除的芐基。示例性陰離子包括但不限于羧酸根(例如乙酸根、苯甲酸根、三氟乙酸根)、鹵素離子(例如、氯離子、溴離子、碘離子)、硫酸根(例如,單硫酸根、硫酸氫根)和磷酸根(例如,單磷酸根、磷酸氫根)。在一些實施方案中,烷基和包含烷基的基團(例如,烷氧基、烷酰基、烷基氨基、氨基烷基、雜烷基)包含1-6個碳原子。在一些實施方案中,芳基和包含芳基的基團(例如,芳酰基)包含6-12個碳原子。在一些實施方案中,雜芳基和包含雜芳基的基團(例如,雜芳酰基)包含1-10個碳原子和1-4個選自氧、氮和硫的雜原子。術語“雜環基”是指雜環基團,并且包括類似于環烷基的閉合的環結構,其中環中的一個或多個,例如,1、2或3個碳原子是除碳之外的元素,例如,氮、硫或氧。雜環基團可以是飽和的或不飽和的。術語雜環基團包括經由至環中的一個雜原子的鍵或至環中的一個碳的鍵連接至核心結構的環。雜環基包括例如哌啶、哌嗪、吡咯烷、嗎啉、內酯、內酰胺等。術語"C3-6雜環基"是指在環中具有3-6個碳的雜環基。在一個示例性實施例中,四氫呋喃基除了氧之外在環中具有四個碳,因此是C4雜環基。如本申請所用,術語“任選被取代的”是指被獨立地選自以下的一個或多個取代基任選取代:例如,烷基、烷氧基、烷基氨基、氨基烷基、羥基、鹵素、氰基、硝基、羧基、羧基烷基、酰胺基團、脲、環烷基、雜環基(例如,雜環烷基)、芳基和雜芳基取代基。6.2縮寫AP=4-氨基苯酚AUC=曲線下面積BOC=Boc、叔丁基氧基羰基或叔丁氧基羰基DAP=4-(二甲基氨基)苯酚DMF=N,N-二甲基甲酰胺EDTA=乙二酸四乙酸ESI=電子噴射電離GC=氣相色譜法HPLC=高效液相色譜法HRMS=高分辨質譜法KTFA=三氟乙酸鉀LOD=干燥失重NMP=N-甲基吡咯烷酮NMR=核磁共振PMB=對-甲氧基芐基、4-甲氧基芐基或MPMPd2dba3=三(二亞芐基丙酮)二鈀(0)TBME=叔丁基甲基醚、甲基叔丁基醚或MTBETDAPP=亞磷酸三(4-(二甲基氨基)苯基)酯THF=四氫呋喃TFA=三氟乙酸6.3制備方法在一個方面中,本申請提供了一種用于制備式(II)的化合物或其鹽的方法,包括如下步驟:在包含(a)鈀源和(b)結合鈀的配體的試劑的存在下使式(III)的化合物或其鹽與親核試劑(Nuc)混合,例如使式(III)的化合物或其鹽與親核試劑(Nuc)反應,以形成式(II)的化合物或其鹽。在一個方面中,本申請提供了一種用于制備式(II)的化合物或其鹽的方法:包括如下步驟:在包含(a)鈀源和(b)結合鈀的配體的試劑的存在下使式(III)的化合物或其鹽與親核試劑(Nuc)混合,例如使式(III)的化合物或其鹽與親核試劑(Nuc)反應:以形成式(II)的化合物或其鹽;其中:Nuc是R4-M,其中M是H、金屬陽離子、非金屬陽離子或孤對電子;R4是由Nuc的添加所產生的基團;LG是選自鹵素或-OC(O)R8的離去基團,其中R8選自C1-6-烷基和鹵代烷基;R1選自:Y選自:R2是氫或烷氧基;R3選自氫和氧保護基;Z’選自氫和氧保護基;R’選自氫和氧保護基;和R”選自氫和氮保護基。在一個實施方案中,所述在包含(a)鈀源和(b)結合鈀的配體的試劑的存在下使式(III)的化合物混合,例如使式(III)的化合物反應的步驟形成鈀pi-烯丙基中間體。在一些實施方案中,式(II)的化合物是式(IIa)的化合物。在一些實施方案中,式(III)的化合物是式(IIIa)的化合物。在另一個方面中,本申請提供了一種用于制備式(IIa)的化合物或其鹽的方法:包括如下步驟:在包含(a)鈀源和(b)結合鈀的配體的試劑的存在下使式(IIIa)的化合物或其鹽與具有式(X)的化合物的結構的親核試劑(Nuc)混合,例如,使式(IIIa)的化合物或其鹽與具有式(X)的化合物的結構的親核試劑(Nuc)反應,所述式(IIIa)為:所述式(X)為:以形成式(IIa)的化合物或其鹽,其中:是選自氯離子、乙酸根、三氟乙酸根和硫酸氫根的陰離子;LG是選自鹵素或-OC(O)R8的離去基團,其中R8選自C1-6-烷基和鹵代烷基;R3是氧保護基;R’是氧保護基;R5是氮保護基;和R6是氮保護基。在一個實施方案中,所述在包含(a)鈀源和(b)結合鈀的配體的試劑的存在下使式(IIIa)的化合物混合,例如使式(IIIa)的化合物反應的步驟形成pi-烯丙基(即,π-烯丙基)中間體。在該方法的另一個實施方案中,所述試劑進一步包含(c)鹽添加劑。在另一個實施方案中,所述鹽添加劑選自鉀鹽、鈉鹽、鋰鹽、銀鹽和銅鹽。在另一個實施方案中,所述鹽添加劑選自三氟乙酸鉀、三氟乙酸鈉、三氟乙酸鋰、三氟甲磺酸鉀、三氟甲磺酸鈉、三氟甲磺酸鋰、三氟甲磺酸銀和硫酸銅。在一個實施方案中,R’是叔丁基;R3是4-甲氧基芐基醚(即,4-甲氧基芐基);R5是叔丁基氧基羰基;和R6是三苯基甲基。在一個實施方案中,LG是鹵素或-OC(O)R8,其中R8選自C1-6-烷基和鹵代烷基。在另一個實施方案中,LG是氯或–OC(O)CF3。在一個實施方案中,所述鈀源選自二(乙腈)二氯化鈀(II)、二(乙酰基丙酮酸)鈀(II)、二(芐腈)二氯化鈀(II)、二(二亞芐基丙酮)鈀、乙酸鈀(II)、三氟乙酸鈀(II)、二氯化鈀(II)、二溴化鈀(II)、四(乙腈)四氟硼酸鈀(II)、三(二亞芐基丙酮)二鈀(0)、三(二亞芐基丙酮)二鈀(0)-氯仿加合物、[1,2-二(二苯基膦基)乙烷]二氯化鈀(II)、1,1’-二(二苯基膦基)二茂鐵-二氯化鈀(II)二氯甲烷加合物、二(三環己基膦)鈀(0)、二(三乙基膦)二氯化鈀(II)、二(三苯基膦)乙酸鈀(II)、二(三苯基膦)二氯化鈀(II)、二(三-叔丁基膦)鈀(0)、二[1,2-二(二苯基膦基)乙烷]鈀(0)、二[三(鄰-甲苯基)膦]二氯化鈀(II)、二氯二(三環己基膦)鈀(II)、四(三苯基膦)鈀(0)、四(三乙基膦)鈀(0)和反式-芐基(氯)二(三苯基膦)鈀(II)。在另一個實施方案中,所述鈀源選自三(二亞芐基丙酮)二鈀(0)、三(二亞芐基丙酮)二鈀(0)-氯仿加合物、四(三苯基膦)鈀、乙酸鈀(II)、三氟乙酸鈀(II)、二氯化鈀(II)、二溴化鈀(II)和二(乙腈)二氯化鈀(II)。在另一個實施方案中,所述鈀源以相對于式(IIIa)的化合物而言約0.5mol%至約5摩爾%的量存在。在一個實施方案中,所述結合鈀的配體是式(VI)的亞磷酸酯配體:其中R7在每次出現時獨立地選自苯基、雜芳基、雜環基和C1-6烷基,其中所述苯基、雜芳基、雜環基和C1-6烷基任選被鹵素、C1-6烷基、C1-6烷氧基或N(R9)2中的一個或多個取代,并且其中所述苯基和雜芳基任選被稠合的C3-6環烷基或C3-6雜環基進一步取代;R8選自苯基、雜芳基、雜環基和C1-6烷基,其中所述苯基、雜芳基、雜環基和C1-6烷基任選被鹵素、C1-6烷基、C1-6烷氧基或N(R9)2中的一個或多個取代,并且其中所述苯基和雜芳基任選被稠合的C3-6環烷基或C3-6雜環基取代,或R8是其中L選自–(CH2)n–、R8任選通過鍵或-(CH2)n-與一個R7連接以形成一個環,或與每個R7連接以形成兩個環;每個R9是C1-6烷基,或兩個R9可以組合形成3-10元雜環基,其中雜環基包含1-3個氮原子并且任選被C1-6烷基或C(O)-C1-6烷基取代;和n是1、2或3。在另一個實施方案中,所述結合鈀的配體是亞磷酸酯配體,其選自:在一個實施方案中,所述結合鈀的配體以相對于鈀源而言約3:1至約10:1的摩爾比存在。在一個實施方案中,本申請提供的方法進一步包括如下步驟:使式(IIa)的化合物或其鹽與強酸混合,例如,使式(IIa)的化合物或其鹽與強酸反應以形成式(Va)的化合物:其中是藥學上可接受的陰離子。所述強酸可以是布郎斯臺德酸(例如,三氟乙酸、鹽酸)或路易斯酸(例如,TiCl4)。在一個具體實施方案中,所述強酸是三氟乙酸。在一些實施方案中,所述方法包括分離式(Va)的化合物的步驟。在一些實施方案中,所述分離步驟包括用非極性溶劑萃取混合物,所述非極性溶劑例如芳族溶劑諸如甲苯、二甲苯或異丙基苯,或烴溶劑諸如戊烷類、己烷類或庚烷類,或其一種或多種的混合物。在一些實施方案中,所述用非極性溶劑萃取混合物在約-25至約-5℃的溫度進行。在一些實施方案中,所述分離步驟包含添加約2至約6體積,例如,約2、約3、約4、約5或約6體積的提供動力學上有利的沉淀條件的溶劑,例如,乙腈。在一些實施方案中,所述分離步驟包含過濾固體以分離式(Va)的化合物。在一些實施方案中,所述式(Va)的化合物具有化合物(VII)的結構:在一些實施方案中,式(II)的化合物,例如,式(IIa)的化合物是式(IIb)的化合物或其鹽。在一些實施方案中,式(III)的化合物,例如,式(IIIa)的化合物是式(IIIb)的化合物或其鹽。在一些實施方案中,親核試劑(Nuc),例如,式(X)的化合物具有親核試劑(UBT)的結構。在另一個方面中,本申請提供了一種用于制備式(IIb)的化合物或其鹽的方法:包括如下步驟:在包含(a)鈀源、(b)結合鈀的配體和(c)鹽添加劑的試劑的存在下使式(IIIb)的化合物或其鹽與親核試劑(UBT)混合,例如,使式(IIIb)的化合物或其鹽與親核試劑(UBT)反應,所述式(IIIb)為:所述親核試劑(UBT)為:以形成式(IIb)的化合物或其鹽,其中:是藥學上可接受的陰離子;所述鈀源選自三(二亞芐基丙酮)二鈀(0)、三(二亞芐基丙酮)二鈀(0)-氯仿加合物、四(三苯基膦)鈀、乙酸鈀(II)、三氟乙酸鈀(II)、二氯化鈀(II)、二溴化鈀(II)和二(乙腈)二氯化鈀(II);所述結合鈀的配體選自且所述鹽添加劑選自三氟乙酸鉀、三氟乙酸鈉、三氟乙酸鋰、三氟甲磺酸鉀、三氟甲磺酸鈉、三氟甲磺酸鋰、三氟甲磺酸銀和硫酸銅。在一個實施方案中,所述方法進一步包括如下步驟:使式(IIb)的化合物或其鹽與強酸混合,例如使式(IIb)的化合物或其鹽與強酸反應以形成式(Va)的化合物:其中是藥學上可接受的陰離子。在本申請中公開的方法的某些實施方案中,具體的反應混合物、試劑和/或溶劑(例如,鈀源、結合鈀的配體、鹽添加劑)以及量或當量或量或當量的范圍選自表1和表2。示例性方法公開在實施例中。在另一個方面中,本申請提供了一種用于制備式(VIII)的化合物或其鹽的方法:包括如下步驟:在包含(a)鈀源和(b)結合鈀的配體的試劑的存在下使式(IX)的化合物或其鹽與親核試劑(Nuc)(例如,R4–M)混合,例如使式(IX)的化合物或其鹽與親核試劑(Nuc)(例如,R4–M)反應:以形成式(VIII)的化合物或其鹽。所述親核試劑、鈀源、結合鈀的配體以及式(VIII)和(IX)的化合物的變量如上面所定義。式(VIII)的化合物可以經由PCT申請PCT/US2014/027706中所述的方法轉化成式(II)的化合物,其由此通過引用以其整體并入。在一些實施方案中,所述式(VIII)的化合物是式(VIIIa)的化合物。在一些實施方案中,所述式(IX)的化合物是式(IXa)的化合物。在另一個方面中,本申請提供了一種用于制備式(VIIIa)的化合物或其鹽的方法:包括如下步驟:在包含(a)鈀源和(b)結合鈀的配體的試劑的存在下使式(IXa)的化合物或其鹽與親核試劑例如式(X)的化合物混合,例如使式(IXa)的化合物或其鹽與親核試劑例如式(X)的化合物反應,所述式(IXa)為:所述式(X)為:以形成式(VIIIa)的化合物或其鹽,其中:是藥學上可接受的陰離子;LG是鹵素、-O(C=O)N(R18)2、-O(C=O)OR18或-OC(O)R18,其中R18選自C1-6烷基和鹵代烷基;R3是氧保護基;R5是氮保護基;和R6是氮保護基。6.3.1溶劑任何不抑制反應的有機溶劑均是合適的溶劑。合適的溶劑包括但不限于:N-甲基吡咯烷酮、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、四氫呋喃、甲基四氫呋喃、甲基叔丁基醚、二噁烷、乙腈、丙酮、二氯甲烷、碳酸丙烯酯、甲醇、乙醇、叔丁醇、叔戊醇及其任何組合。在一些實施方案中,所述溶劑是四氫呋喃。在一些實施方案中,所述溶劑是四氫呋喃和N-甲基吡咯烷酮的混合物。6.3.2溫度如本申請所述的用于制備式(II)的化合物,例如式(IIa)的化合物的方法可以在一定溫度范圍內進行。在一些實施方案中,形成式(II)的化合物,例如式(IIa)的化合物的溫度是在約0至約65℃,諸如約10至約40、約15至約35、約20至約30或約23至約30℃的范圍內。在一些實施方案中,形成式(II)的化合物,例如式(IIa)的化合物的溫度是在約0、約10、約20、約25、約30、約40、約50或約60℃。在一些實施方案中,所述溫度是在約23至約30℃范圍內的環境溫度。6.3.3時間如本申請所述的用于制備式(II)的化合物,例如式(IIa)的化合物的方法可以在各種時間期間內進行。在一些實施方案中,完成式(II)的化合物,例如式(IIa)的化合物的形成的反應時間是在約10min至約7天,諸如約10min至約2天、約15min至約24小時、約30min至約12小時、約1至約6小時或約2至約5小時的范圍內。在一些實施方案中,完成式(II)的化合物,例如式(IIa)的化合物的形成的反應時間是約0.25、約0.5、約1、約2、約4、約6、約8、約12或約24小時。在一些實施方案中,所述反應時間是約15min。在一些實施方案中,所述反應時間是約4小時。6.3.4鈀的去除反應完成之后,可以通過本領域已知的任何方法從反應混合物中去除鈀,包括但不限于吸附、萃取和結晶方法。在一個示例性實例中,可以將反應后溶液中的鈀吸附到活性炭上,然后經珍珠巖過濾溶液以從期望的有機產物中去除鈀。在另一個實例中,可以用清除樹脂,例如,固定在固體載體上的硫脲,例如TU(JohnsonMattheyFinlandOy)處理反應混合物,然后過濾以除去鈀物質。在一些實施方案中,在鈀-介導的偶聯后,使反應混合物經歷酸性水洗。在一些實施方案中,所述酸性水洗具有在約0.5至約2.5范圍內的pH。洗滌的主要目的是從工藝流中去除鈀。該水性廢物流含有大量在所述方法中使用的鈀。在某些實施方案中,用二硫代氨基甲酸鹽,例如二乙基二硫代氨基甲酸鈉或吡咯烷二硫代氨基甲酸銨處理工藝流,以用于提供本公開的化合物,例如具有0.001-10ppm的鈀水平的式(II)的化合物(諸如式(IIa)的化合物、式(IIb)的化合物)或式(V)的化合物(諸如式(Va)的化合物,例如,頭孢洛扎硫酸鹽)。通常,本公開的化合物,例如頭孢洛扎TFA含有約100ppm鈀。為了將鈀水平降低至藥學上可接受的水平,將本公開的化合物,例如頭孢洛扎TFA首先溶解在水性介質中并用約0.1至約10摩爾%的二硫代氨基甲酸鹽,例如二乙基二硫代氨基甲酸鈉或二甲基二硫代氨基甲酸鈉處理,得到含有鈀的固體的精細漿料。將這些含有鈀的固體從批料中濾除,所述批料通過先前報道的順序經歷鹽交換以提供具有0.001-10ppm殘余鈀的式(Va)的化合物,例如頭孢洛扎硫酸鹽。在一些實施方案中,所述鈀的去除包括順序進行的一個或多個,例如兩個、三個、四個或五個方法,以提供含有殘余量的鈀的本公開的化合物。使用的方法可以是重復一次或多次,例如兩次、三次、四次或五次的相同方法或者是不同方法的組合。不同方法的組合可以以任何順序進行。在一個示例性實例中,酸性水洗(pH1)來自鈀反應的有機反應混合物去除約90%反應中使用的鈀。在另一個示例性實例中,酸性水洗(pH1),隨后用二硫代氨基甲酸鹽,即吡咯烷二硫代氨基甲酸銨(APDTC)處理并經0.4μm過濾器過濾析出的固體,得到包含本公開的化合物,例如式(II)的化合物(諸如式(IIa)的化合物、式(IIb)的化合物)或式(V)的化合物(諸如式(Va)的化合物)以及<10ppm鈀的有機溶液。在本申請所述的方法之后,本公開的化合物,例如式(II)的化合物(諸如式(IIa)的化合物、式(IIb)的化合物)或式(V)的化合物(諸如式(Va)的化合物)含有殘余水平的鈀。在一些實施方案中,所述鈀以如下范圍存在:約0.01百萬分率(ppm)至約50ppm鈀,諸如約0.01至約40、約0.01至約30、約0.01至約10、約0.01至約5、約0.01至約3、約0.01至約2、約0.01至約1.5、約0.01至約1、約0.01至約0.5;約0.02至約40、約0.02至約30、約0.02至約10、約0.02至約5、約0.02至約3、約0.02至約2、約0.02至約1.5、約0.02至約1、約0.02至約0.5;約0.05至約40、約0.05至約30、約0.05至約10、約0.05至約5、約0.05至約3、約0.05至約2、約0.05至約1.5、約0.05至約1、約0.05至約0.5;約0.1至約40、約0.1至約30、約0.1至約10、約0.1至約5、約0.1至約3、約0.1至約2、約0.1至約1.5、約0.1至約1、約0.1至約0.5;約0.2至約40、約0.2至約30、約0.2至約10、約0.2至約5、約0.2至約3、約0.2至約2、約0.2至約1.5、約0.2至約1、約0.2至約0.5;約0.5至約40、約0.5至約30、約0.5至約10、約0.5至約5、約0.5至約3、約0.5至約2或約0.5至約1.5ppm鈀。在一些實施方案中,所述鈀以約0.01、約0.02、約0.05、約0.1、約0.2、約0.3、約0.4、約0.5、約1、約1.5、約2、約5、約10、約20或約50ppm鈀存在。6.3.5鈀的回收鈀是昂貴的稀土金屬。可以將由本公開的方法回收的用過的、粗制的鈀混合物銷售給金屬處理商進行重復利用。鈀的回收支付包含鈀的大規模制造方法的總成本是合乎需要的,該成本涉及鈀催化劑的購買和含有鈀的廢物流的處置中的成本二者。在一些情況下,鈀回收方法對于本公開的方法的經濟性是關鍵的。在一些實施方案中,在鈀反應完成后回收鈀,用于重復利用和加工。在一些實施方案中,在鈀-介導的偶聯以形成式(II)的化合物,例如式(IIa)的化合物例如式(IIb)的化合物之后,使反應混合物進行約0.5至約2.5的pH范圍內的酸性水洗,其主要目的是從工藝流中去除鈀。在一些實施方案中,酸性水溶液的pH從約0.5至約5.5范圍內的初始pH升高至約5至約10范圍內的最終pH。在一些實施方案中,添加氧化劑,例如漂白劑。回收方法導致含有固體鈀的固體析出,其可以從溶液中濾除。在一個實例中,蒸餾(夾套溫度=40℃)合并的酸性水層(pH范圍為0.5-2.5),直到去除約10%的體積(去除的溶劑大部分為THF)。將溶液用NH4OH處理,直到pH為約5至約10(目標pH為約7.5),同時保持溫度低于20℃。這導致含有白色固體的深色漿料。之后,將批料用0.4當量(相對于批料的TATD-CLE投料量)的NaClO(即,漂白劑;0.1-2當量,目標為相對于TATD-CLE投料量而言約0.4當量)處理。溶液立即變紅,并在室溫攪拌2小時。然后,將纖維素加入至批料中,并將漿料經纖維素墊過濾,提供富集鈀的濾餅。回收最初在水溶液中存在的約90%的鈀或最初在方法中使用的約80%的鈀。6.4組合物在一個方面中,本申請提供了根據本申請所述的任一方法制備的組合物,其包含式(Va)的化合物,其中是藥學上可接受的陰離子。在一些實施方案中,是氯離子、溴離子、碘離子、硫酸根、甲苯磺酸根、甲烷磺酸根、乙二磺酸根、馬來酸根、磷酸根、酮戊二酸根、三氟乙酸根或三氟甲烷磺酸根。在一些優選的實施方案中,是硫酸根。在一些實施方案中,所述式(Va)的化合物具有化合物(VII)的結構:在一個方面中,本申請提供了根據本申請所述的任一方法制備的組合物,其包含式(Vb)的化合物,其中是藥學上可接受的陰離子。在一些實施方案中,所述組合物(例如包含式(Va)的化合物或式(Vb)的化合物)包含鈀。在一些實施方案中,所述鈀以如下范圍存在:約0.01百萬分率(ppm)至約50ppm鈀,諸如約0.01至約40、約0.01至約30、約0.01至約10、約0.01至約5、約0.01至約3、約0.01至約2、約0.01至約1.5、約0.01至約1、約0.01至約0.5;約0.02至約40、約0.02至約30、約0.02至約10、約0.02至約5、約0.02至約3、約0.02至約2、約0.02至約1.5、約0.02至約1、約0.02至約0.5;約0.05至約40、約0.05至約30、約0.05至約10、約0.05至約5、約0.05至約3、約0.05至約2、約0.05至約1.5、約0.05至約1、約0.05至約0.5;約0.1至約40、約0.1至約30、約0.1至約10、約0.1至約5、約0.1至約3、約0.1至約2、約0.1至約1.5、約0.1至約1、約0.1至約0.5;約0.2至約40、約0.2至約30、約0.2至約10、約0.2至約5、約0.2至約3、約0.2至約2、約0.2至約1.5、約0.2至約1、約0.2至約0.5;約0.5至約40、約0.5至約30、約0.5至約10、約0.5至約5、約0.5至約3、約0.5至約2或約0.5至約1.5ppm鈀。在一些實施方案中,所述鈀以約0.01、約0.02、約0.05、約0.1、約0.2、約0.3、約0.4、約0.5、約1、約1.5、約2、約5、約10、約20或約50ppm鈀存在。在一些優選的實施方案中,所述組合物中鈀的水平小于鈀的藥學上可接受的水平,例如如在2013年2月1日發行的UnitedStatesPharmacopeia(USP)GeneralChapter<232>ElementalImpurities–Limits,RevisionBulletin中所規定的水平:表A:藥品的元素狀態雜質(從在2013年2月1日發行的USPChapter<232>RevisionBulletin中摘錄)aPDE=基于50-kg人的可允許的日暴露。bLVP=大體積腸胃外。表B:藥物物質和賦形劑的默認濃度限值(從在2013年2月1日發行的USPChapter<232>RevisionBulletindatedFebruary1,2013中摘錄)在一些實施方案中,確定在任一本申請所述的藥物組合物中的鈀的水平。例如當所述組合物以單位劑型的形式,例如與他佐巴坦組合的形式,例如以的形式配制時,可以測量鈀的水平。在另一個方面中,本申請提供了組合物,其包含:(a)式(IIIb)的化合物:或其鹽,(b)式(UBT)的親核試劑:(c)鈀源;(d)結合鈀的配體;和(e)鹽添加劑。在一個實施方案中,所述鈀源選自三(二亞芐基丙酮)二鈀(0)、三(二亞芐基丙酮)二鈀(0)-氯仿加合物、四(三苯基膦)鈀、乙酸鈀(II)、三氟乙酸鈀(II)、二氯化鈀(II)、二溴化鈀(II)和二(乙腈)二氯化鈀(II)。在一個實施方案中,所述結合鈀的配體選自在一個實施方案中,所述鹽添加劑選自三氟乙酸鉀、三氟乙酸鈉、三氟乙酸鋰、三氟甲磺酸鉀、三氟甲磺酸鈉、三氟甲磺酸鋰、三氟甲磺酸銀和硫酸銅。在另一個實施方案中,所述組合物進一步包含pi-烯丙基中間體。在另一個實施方案中,所述組合物進一步包含式(IIb)的化合物:其中是陰離子,例如藥學上可接受的陰離子。在另一個實施方案中,所述鹽添加劑選自三氟乙酸鉀、三氟乙酸鈉和三氟乙酸鋰,并且所述組合物進一步包含式(IIIc)的化合物6.4.1藥物組合物通過本公開的方法制備的化合物,例如式(Va)的化合物,例如化合物(VII),可以被配制成藥物組合物。所述藥物組合物可以任選進一步包含β-內酰胺酶抑制劑諸如他佐巴坦。所述藥物組合物可以通過本申請所述的方法來獲得。特別地,藥物組合物可以通過包括如下步驟的方法來獲得:形成含有本公開的化合物,例如式(Va)的化合物的水溶液,并凍干所述水溶液以獲得藥物組合物。所述水溶液可以額外地包含賦形劑、穩定劑、pH調節添加劑(例如緩沖劑)等。這些添加劑的非限制性例子包括氯化鈉、檸檬酸和L-精氨酸。例如使用氯化鈉可以導致更大的穩定性;L-精氨酸可以用于調節pH并增加頭孢洛扎的溶解度;且檸檬酸由于其螯合金屬離子的能力而可以用于防止產物的變色。特別地,所述水溶液可以包含頭孢洛扎硫酸鹽和額外的組分諸如氯化鈉以穩定頭孢洛扎,以及包含堿化劑諸如L-精氨酸以在凍干之前提供約5-7的pH。所述藥物組合物可以被凍干(冷凍干燥)并作為凍干物貯存以用于隨后的重構。與藥物制劑的凍干有關的示例性公開包括Konan等人,Int.J.Pharm.2002233(1-2),293-52;Quintanar-Guerrero等人,J.Microencapsulation199815(1),107-119;Johnson等人,J.PharmaceuticalSci.2002,91(4),914-922;和Tang等人,PharmaceuticalRes.2004,21(4),191-200;其公開內容通過引用并入本申請。作為凍干的替代方式,可以將藥物組合物噴霧干燥或冷凍貯存,然后在給藥前解凍、重構和稀釋。在一些實施方案中,將本公開的藥物組合物,例如包含式(Va)的化合物,例如化合物(VII)配制成藥學上可接受的鹽。術語“藥學上可接受的鹽”是指式(Va)的化合物,例如化合物(VII)的相對無毒的、無機的和有機的酸加成鹽。這些鹽可以在化合物的最終分離和純化的過程中原位制備,或可以通過單獨地混合以其游離堿形式存在的純化的化合物與合適的有機或無機酸,例如使以其游離堿形式存在的純化的化合物與合適的有機或無機酸反應并分離由此形成的鹽。代表性的鹽包括溴化物、氯化物、硫酸鹽、硫酸氫鹽、磷酸鹽、硝酸鹽、乙酸鹽、戊酸鹽、油酸鹽、棕櫚酸鹽、硬脂酸鹽、月硅酸鹽、苯甲酸鹽、乳酸鹽、磷酸鹽、甲苯磺酸鹽、檸檬酸鹽、馬來酸鹽、富馬酸鹽、琥珀酸鹽、酒石酸鹽、萘甲酸鹽、甲磺酸鹽、葡庚糖酸鹽、乳糖酸鹽、月桂基磺酸鹽和氨基酸鹽等。參見,例如Berge等人1977,“PharmaceuticalSalts,”J.Pharm.Sci.66:1-19。可以通過混合具有期望的純度的藥物活性成分與任選的本領域中通常使用的藥學上可接受的載體、賦形劑或穩定劑(其在本申請中全部被稱為“載體”),即,緩沖劑、穩定劑、防腐劑、等滲劑、非離子洗滌劑、抗氧化劑及其它多種添加劑將本公開的頭孢菌素化合物,例如式(Va)的化合物,例如化合物(VII)的藥物組合物制備成凍干制劑或者水性溶液來來貯存。參見,Remington’sPharmaceuticalSciences,第16版(Osol編1980)。在一些實施方案中,使用約2mM至約50mM的量的緩沖劑來幫助維持pH在大約生理條件下的范圍。用于本公開的合適的緩沖劑包括有機和無機酸及其鹽,諸如檸檬酸鹽緩沖劑(例如檸檬酸單鈉-檸檬酸二鈉混合物、檸檬酸-檸檬酸三鈉混合物、檸檬酸-檸檬酸單鈉混合物等)、琥珀酸鹽緩沖劑(例如琥珀酸-琥珀酸單鈉混合物、琥珀酸-氫氧化鈉混合物、琥珀酸-琥珀酸二鈉混合物等)、酒石酸鹽緩沖劑(例如酒石酸-酒石酸鈉混合物、酒石酸-酒石酸鉀混合物、酒石酸-氫氧化鈉混合物等)、富馬酸鹽緩沖劑(例如富馬酸-富馬酸單鈉混合物、富馬酸-富馬酸二鈉混合物、富馬酸單鈉-富馬酸二鈉混合物等)、葡糖酸鹽緩沖劑(例如葡糖酸-葡糖酸鈉混合物、葡糖酸-氫氧化鈉混合物、葡糖酸-葡糖酸鉀混合物等)、草酸鹽緩沖劑(例如草酸-草酸鈉混合物、草酸-氫氧化鈉混合物、草酸-草酸鉀混合物等)、乳酸鹽緩沖劑(例如乳酸-乳酸鈉混合物、乳酸-氫氧化鈉混合物、乳酸-乳酸鉀混合物等)和乙酸鹽緩沖劑(例如乙酸-乙酸鈉混合物、乙酸-氫氧化鈉混合物等)。此外,可以使用磷酸鹽緩沖劑、組氨酸緩沖劑和三甲基胺鹽諸如Tris。在一些實施方案中,以0.01%-1%(w/v)的量添加防腐劑。本公開使用的合適的防腐劑包括苯酚、芐醇、間甲酚、尼泊金甲酯、尼泊金丙酯、十八基二甲基芐基氯化銨、苯扎鹵銨(例如苯扎氯銨、苯扎溴銨和苯扎碘銨)、氯化六甲季銨和尼泊金烷基酯諸如尼泊金甲酯或丙酯、兒茶酚、間苯二酚、環己醇和3-戊醇。在一些實施方案中,加入有時被稱為“穩定劑”的等滲劑以確保本公開的液體組合物的等滲性,并且其包括多羥基糖醇,例如三羥基或更高的糖醇,諸如甘油、赤蘚醇、阿糖醇、木糖醇、山梨醇和甘露醇。穩定劑是指廣泛類別的賦形劑,其在功能上包括從增量劑至增溶治療劑或幫助防止變性或粘附至容器壁的添加劑。典型的穩定劑可以為多羥基糖醇(上文列舉的);氨基酸諸如精氨酸、賴氨酸、甘氨酸、谷氨酰胺、精氨酸、組氨酸、丙氨酸、鳥氨酸、L-亮氨酸、2-苯基丙氨酸、谷氨酸、蘇氨酸等、有機糖或糖醇,諸如乳糖、海藻糖、水蘇糖、甘露醇、山梨醇、木糖醇、核糖醇、Myo-肌醇、半乳糖醇、甘油等,包括環醇諸如肌醇;聚乙二醇;氨基酸聚合物;含硫的還原劑,諸如硫脲、谷胱甘肽、硫辛酸、巰基乙酸鈉、硫代甘油、α-二羥基丙硫醇和硫代硫酸鈉;低分子量多肽(例如10個殘基或更少的肽);蛋白質諸如人血清白蛋白、牛血清白蛋白、明膠或免疫球蛋白;親水性聚合物,諸如聚乙烯吡咯烷酮、單糖,諸如木糖、甘露糖、果糖、葡萄糖;二糖諸如乳糖、麥芽糖、蔗糖和三糖諸如棉籽糖;和多糖諸如葡聚糖。在一些實施方案中,穩定劑以0.1-10,000重量/藥物活性成分的重量份的范圍存在。組合物將通常作為無菌藥物組合物的一部分供應,該組合物通常包括藥學上可接受的載體。該組合物可以為任何合適的形式(取決于期望的給藥方法)。例如可以將藥物組合物配制成水溶液并通過靜脈內注射或靜脈內輸注給藥。藥物組合物可以包括頭孢洛扎鹽,例如化合物(VII)(通過本申請所述的方法獲得),聯合β-內酰胺酶抑制劑,諸如他佐巴坦(CAS#:89786-04-9),阿維巴坦(CAS#1192500-31-4)、舒巴克坦(CAS#68373-14-8)和/或克拉維酸(CAS#58001-44-8)。β-內酰胺酶抑制劑可以以晶體或無定形,諸如凍干的他佐巴坦或結晶的他佐巴坦形式(例如US專利8,476,425和5,763,603)包括以獲得所述藥物組合物。可以配制包含式(Va)的化合物,例如化合物(VII)的藥物組合物以通過腸胃外(包括皮下、肌內和靜脈內)給藥治療感染。在一個具體實施方案中,將本申請所述的藥物組合物配制成通過靜脈內注射或輸注給藥。呈凍干單位劑型形式(例如小瓶中的粉末)的藥用抗生素組合物可以包括頭孢洛扎硫酸鹽和穩定量的氯化鈉(例如125-500mg氯化鈉/1,000mg頭孢洛扎活性物)。單位劑型可以用藥學上可接受的載體溶解,然后靜脈內給藥。在另一個方面中,藥用抗生素組合物可以包括通過包括如下步驟的方法獲得的頭孢洛扎硫酸鹽:凍干含有頭孢洛扎和穩定量的氯化鈉的水溶液,其中穩定量的氯化鈉為凍干前的水溶液中約125-500mg氯化鈉/1,000mg頭孢洛扎活性物。6.5治療方法在一個方面中,本申請提供了用于在哺乳動物中治療細菌感染的方法,其包括對所述哺乳動物給藥治療有效量的藥物組合物,所述藥物組合物包含根據本申請所述的一種或多種方法制備的式(Va)的化合物,例如化合物(VII)。用于治療哺乳動物中的細菌感染的方法可以包括給所述哺乳動物給藥治療有效量的包含頭孢洛扎硫酸鹽和氯化鈉的藥物組合物。如本申請所用,“哺乳動物”可以為任何哺乳動物,諸如小鼠、大鼠、狗、貓、馬、豬、牛或靈長類動物,諸如人。在某些實施方案中,所述哺乳動物是人。所述哺乳動物可以成人或青少年。式(Va)的化合物,例如化合物(VII)的藥物組合物可以與甲硝唑組合使用來治療由諸如下述革蘭氏陰性和陽性微生物導致的復雜的腹內感染:大腸桿菌(包括產CTX-M-14/15ESBLs的菌株)、肺炎克雷伯菌(包括產CTX-M-15ESBLs的菌株)、銅綠假單胞菌、陰溝腸桿菌、產酸克雷伯菌、奇異變形桿菌、脆弱擬桿菌、卵形擬桿菌、多形擬桿菌、普通擬桿菌、咽峽炎鏈球菌、星座鏈球菌和唾液鏈球菌。藥物組合物可以用于治療復雜的尿道感染,包括由下述革蘭氏陰性微生物導致的具有或不具有并存的菌血癥的腎盂腎炎:大腸桿菌(包括對左氧氟沙星耐藥和/或產生CTX-M-14/15ESBLs的菌株)、肺炎克雷伯菌(包括對左氧氟沙星耐藥和/或產生CTX-M-15ESBLs的菌株)、奇異變形桿菌和銅綠假單胞菌。包含通過本申請公開的一種或多種方法制備的式(Va)的化合物,例如化合物(VII)和他佐巴坦(其量為提供1g頭孢洛扎活性物/500mg他佐巴坦酸)的藥物組合物的推薦劑量方案為在≥18歲的患者中每8小時經1小時通過靜脈內(IV)輸注給藥1.5g。治療的持續時間應當由感染的嚴重度和部位以及患者的臨床和細菌學進展來指引(例如對于復雜的腹內感染,在4-14天中每8小時,以及對于復雜的尿道感染,包括腎盂腎炎而言,在7天中每8小時)。7.實施例為了舉例說明的目的并且為了描述本發明的某些特定實施方案而在下面給出實施例。然而,本申請中給出的實施例不以任何方式限制權利要求的范圍。對公開的實施方案的各種改變和修飾對于本領域技術人員而言是顯而易見的,并且這樣的改變和修飾可以在不偏離本發明的精神和權利要求的范圍的情況下做出。7.1實施例1:經由鈀催化偶聯TATD-CLE和UBT將TATD-CLE(化合物3,圖1A)(5g毛重,92.3%效能,4.615g活性物,6.775mmol)、UBT(化合物4,圖1A和1B)(4.029g,7.453mmol)和三氟乙酸鉀(2.061g,13.55mmol)加入到反應容器中。然后添加THF(39.23mL),形成不透明的、白色的混懸液。在30℃將反應混合物攪拌30分鐘,保持為白色混懸液。添加亞磷酸三(4-(二甲基氨基)苯基)酯(238mg,0.542mmol),隨后添加Pd2dba3(62mg,0.068mmol)。將反應容器抽真空并用氮氣回填兩次,使溶劑在過程中沸騰。攪拌后,反應混合物變深紫色,然后是淺綠黃色,這指示存在活性催化劑。每小時通過HPLC取樣監測反應進程。在該催化劑負荷的典型的反應時間為約3.5-4小時,其如下定義:相對于TATD-QUATE而言少于2%的剩余的TATD-CLE和/或TATD-TFA。通過上述方法的TATD-QUATE的收率為約92-96%。通過圖1A(化合物5a)中所示的方法的TATD-QUATE的收率為約63-66%。在上述實施例中,形成了短暫存在的式(IIIc)的中間體(在本申請中也成為“TATD-TFA”)。更具體而言,TATD-CLE被部分地轉化為TATD-TFA。然而,TATD-CLE和TATD-TFA都被轉化為TATD-QUATE。7.2實施例2:經由鈀催化偶聯SCLE和UBT在與實施例1中所述類似的偶聯條件下形成SQUATE。7.3實施例3:催化劑的篩選篩選了若干金屬絡合物用于公開的方法。在公開的方法中表現不佳的金屬絡合物包括Fe2(CO)9、Rh(PPh3)3Cl、NiCl2(PPh3)2、CuSO4、AgOTf、Ni(acac)2、Pt(PPh3)2(H2CCH2)、Ru4(C10H15)4Cl4和Ir2(cod)4Cl2。7.4實施例4:合成亞磷酸三(4-(二甲基氨基)苯基)酯(TDAPP),一種結合鈀的配體步驟1:制備4-(二甲基氨基)苯酚(DAP)將4-氨基苯酚(21.8g,200mmol,1.0當量)加入至1L3-頸圓底燒瓶中并將溫度調節至20℃(約15至20℃)。在約22℃(約20至25℃)將甲醇(218mL,10體積)加入至燒瓶中并攪動15min。4-氨基苯酚并沒有完全溶解在MeOH中,由此產生混懸液。緩慢加入H3PO4(85%,2.3g,20mmol,0.10當量),同時維持批料溫度在<25℃。將批料溫度調節至約12℃(10至15℃);然后緩慢加入甲醛(36-38%水溶液,45.9mL,300mmol,3.0當量),同時維持批料溫度在<20℃。在約22℃(20至25℃)將批料攪動30min,得到澄清的淺黃色溶液。然后將溶劑用氮氣脫氣,并用氮氣填充反應器。將Pd/C(10%在活性炭上,4.4g,0.2wt.當量)加入至批料中。將批料在1atm氫氣壓(使用氣囊)在20至25℃劇烈攪動三小時。取一等分試樣的反應混合物(10μL)用于HPLC分析。當氨基苯酚(AP)的量≤2%(相對于DAP而言)時,認為反應完成。反應通常耗時3-5小時,但可以在20至25℃攪拌過夜而沒有觀察到降解。將反應混合物經硅藻土墊過濾(21.8g,1.0wt.當量)。用甲醇(43.2mL,2體積)洗滌硅藻土墊。通過蒸餾將批次濃縮成4體積(86.2mL)。加入甲苯(174.4mL,8體積),隨后添加水(130.8mL,6體積)。然后緩慢加入10NNaOH水溶液(3mL,30mmol,0.15當量),并將批料攪動30min。分離各相,并在真空下濃縮上層有機相為6體積(130.8mL)。加入甲苯(86.4mL,4體積)并通過蒸餾將批次濃縮為8體積(174.4mL)。取一等分試樣的反應混合物(10μL)用于分析甲醇和水的水平。步驟2:制備亞磷酸三(4-(二甲基氨基)苯基)酯(TDAPP)將Et3N(39.03mL,280mmol,1.40當量)加入步驟1中獲得的反應混合物中。將批料溫度調節至2℃(0至5℃)。將PCl3(7.00mL,80mmol,0.40當量)在甲苯(43.2mL,2體積)中的溶液緩慢加入至反應器中,同時保持反應溫度<12℃。反應為放熱的并且混合物變稠。有效的混合需要劇烈攪拌。在15℃(10至20℃)將批料攪動30分鐘。取一等分試樣的反應混合物用于HPLC分析。當相對于TDAPP而言DAP的量≤2%認為反應完成。加入TBME(174.4mL,8體積)并將批料在23℃(20至25℃)攪動2小時以確保從溶液中完全沉淀Et3N·HCl。批料經硅藻土墊(21.8g,1wt.當量)/硅膠(10.8g,0.5wt.當量)過濾。將硅藻土墊/硅膠用TBME(43.2mL,2體積)洗滌。可在濾液中形成白色結晶(Et3N·HCl鹽)的沉淀。如果發生沉淀,則應當經硅藻土墊/硅膠過濾濾液。在43℃(40至45℃)將批料減壓濃縮至3體積(65.4mL)。將2-甲基-2-丁醇(174.4mL,8體積)加入至批料中。在添加醇的過程中可形成沉淀。然后減壓濃縮批料至6體積(130.8mL)。隨后,將2-甲基-2-丁醇(43.2mL,2體積)加入至批料中。在添加2-甲基-2-丁醇后形成稠厚漿料。取一等分試樣的批料以通過GC-FID確定殘余的甲苯含量。甲苯含量可影響后續的結晶。將批料溫度調節至63℃(60至65℃),得到澄清溶液。然后將批料溫度調節至40-45℃并用TDAPP晶種(200mg)種晶。隨后,將批料冷卻至20-25℃并攪動2小時。將批料過濾并將濕濾餅用冷2-甲基-2-丁醇(43.2mL,2體積)洗滌。將批料減壓干燥以得到TDAPP,為白色固體。取一份樣品以通過GC監測干燥。當2-甲基-2-丁醇的量≤5,000ppm時認為批料已干。步驟1和2的總收率為17.5g(理論的60%),分離為白色至淺黃色固體。總純度為97.8%AUC(通過HPLC分析)并且結構通過1HNMR證實。7.5實施例5:由TATD-CLE制備頭孢洛扎TFA7.5.1制備TATD-QUATE7.5.1(a)儀器使用兩個1L夾套玻璃底-排液反應器和內部溫度探針。7.5.1(b)制備TATD-QUATE的材料加料和反應參數表1.制備TATD-QUATE過程中使用的材料方法步驟材料MW(g/mol)當量或體積使用的量1THF72.117.0體積420.0mL2UBT540.661.1mol當量52.4g2TATD-CLE*681.181.0mol當量66.6g2KTFA152.111.5mol當量20.1g2TDAPP439.490.08體積3.10g3THF72.110.5體積30.0mL5Pd2dba3915.700.01mol當量0.81g6THF72.111.0體積60.0mL10EtOAc88.116.0體積360.0mL11水18.0214.0體積600.0mL11NaHSO4120.060.5wt.當量42.0g18Harborlite800114.020.08wt.當量4.8g19EtOAc88.110.5體積30.0mL*將60.0克的TATD-CLE活性量(90.1%效能)用于所有體積和摩爾當量計算。7.5.1(c)制備TATD-QUATE的分步過程1.在15至25℃加入THF[四氫呋喃](373.5g,420.0mL,7.0體積)至反應器。2.加入UBT(52.4g,96.9mmol,1.1當量)、TATD-CLE(66.6g,88.1mmol,1.0當量)、KTFA[三氟乙酸鉀](20.1g,132.1mmol,1.5當量)和TDAPP[亞磷酸三(4-(二甲基氨基)苯基)酯](3.10g,7.0mmol,0.08當量)至反應器1。3.通過洗球加入THF[四氫呋喃](26.7g,30.0mL,0.5體積)至反應器1。4.調節批料溫度至30℃并攪動批料30分鐘。5.加入TDAPP[亞磷酸三(4-(二甲基氨基)苯基)酯](3.10g,7.0mmol,0.08當量)和隨后的Pd2dba3[三(二亞芐基丙酮)二鈀(0)](0.81g,0.9mmol,0.01當量)至批料。6.通過洗球加入THF[四氫呋喃](53.4g,60.0mL,1.0體積)至反應器1并在30℃攪拌批料。7.在4小時后收集樣品,并就反應完成情況對其進行分析。當相對于TATD-QUATE而言剩余≤2.0%TATD-CLE+TATD-TFA時,認為反應已完成(參見,例如圖2中所附的HPLC跡線圖)。8.如果相對于TATD-QUATE而言剩余>2.0%TATD-CLE+TATD-TFA時,則應當將批料攪拌額外的0.5-1小時并重復步驟7。7.5.1(d)淬滅和水性后處理TATD-QUATE的分步操作9.將反應器1中的批料冷卻至5至15℃。10.加入EtOAc[乙酸乙酯](322.9g,360.0mL,6.0體積)至反應器1中的批料中,同時維持批料溫度在2至15℃,然后冷卻至2-8℃。11.如下在反應器2中制備5.0%(w/v)硫酸氫鈉[NaHSO4]的水溶液:首先在水(420.0mL,7.0體積)中溶解(21.0g)NaHSO4,然后調節溶液溫度為2至8℃。12.將反應器1中的批料加入至步驟11中制備的5%NaHSO4溶液中并攪拌批料25至35分鐘,同時維持批料溫度在2至8℃(通過步驟18)。13.停止攪拌并使各相分離至少20分鐘。14.分離下層水層并將其轉移至貯槽中用于處置。15.如下在反應器1中制備5.0%(w/v)硫酸氫鈉[NaHSO4]的水溶液:首先在水(420.0mL,7.0體積)中溶解(21.0g)NaHSO4,然后調節溶液溫度為2至8℃。16.將反應器1中的水溶液(步驟15)加入至反應器2中的批料中并攪拌批料25至35分鐘。17.停止攪拌并使各相分離20至40分鐘。18.分離下層水層并將其轉移至貯槽中用于處置。19.經Harborlite800(4.8g,0.08wt.當量)或相當的過濾材料過濾有機層以去除鈀(0)。20.用EtOAc(26.9g,30.0mL,0.5體積)洗滌Harborlite并合并反應器2中的有機層。7.5.2制備和分離頭孢洛扎TFA7.5.2(a)制備頭孢洛扎TFA(頭孢洛扎三氟乙酸鹽)的材料加料和反應參數表2:制備頭孢洛扎TFA粗制物中使用的材料方法步驟材料MW(g/mol)摩爾當量或體積使用的量22茴香醚108.141.5體積90.0mL25CF3CO2H136.014.5體積270.0mL29甲苯92.1410.0體積600.0mL33ACN41.051.5體積75.0mL34MTBE88.1510.0體積600.0mL36,37MTBE88.155.0體積300.0mL7.5.2(b)制備頭孢洛扎TFA的分步過程21.通過真空蒸餾將反應器2中的批料的體積減少至4.0體積(240.0mL),同時在蒸餾期間維持批料溫度在<20℃。22.加入茴香醚(89.6g,90.0mL,1.5體積)至反應器2中。23.通過真空蒸餾將反應器2中的批料體積減少至3.5體積(210.0mL),同時維持批料溫度在<20℃。24.冷卻批料至<10℃。25.緩慢加入CF3CO2H(402.0g,240.0mL,4.5體積),同時維持批料溫度在<20℃。26.在18至22℃攪拌批料4-10小時。應當連續監測溫度。推薦的監測間隔是1小時。27.4小時后收集一份樣品,并針對反應完成情況對其進行分析。當相對于頭孢洛扎而言剩余≤2.0%頭孢洛扎叔丁基酯時,認為反應已完成(參見,例如圖4)。28.如果頭孢洛扎叔丁基酯>2.0%,則繼續反應0.5-1小時并重復步驟27。29.加入甲苯(522.0g,600.0mL,10.0體積)至反應器2中并調節批料溫度至-15℃。30.在-15℃攪拌混合物30至40分鐘。31.停止攪拌并使各相分離至少20分鐘。32.收集反應器1中的粘性下層相。33.加入ACN[乙腈](71.1g,90.0mL,1.5體積)至反應器1中并調節批料溫度至10至20℃。34.經30至60分鐘(10-20體積/h)加入MTBE[甲基叔丁基醚](444.2g,600.0mL,10.0體積)至反應器1中,同時維持批料溫度在15℃。35.在15℃攪拌所得漿料2-4小時。36.過濾漿料并用MTBE(111.1g,150.0mL,2.5體積)洗滌濾餅。37.用MTBE(111.1g,150.1mL,2.5體積)第二次洗滌濾餅。38.在氮氣流下減壓干燥濾餅。當最終的LOD[干燥失重]值≤19%時,認為干燥過程已完成。通常,干燥的批料具有7.0至9.0%的最終LOD。如本申請所用,“叔丁基頭孢洛扎”、“叔丁基頭孢洛扎酯”或“叔丁基頭孢洛扎”是指式(INT-Va)的化合物:其中是藥學上可接受的陰離子。可能由于在鈀-介導的偶聯反應中實現的收率的顯著增加,先前開發的分離頭孢洛扎TFA的方法沒有提供可分離的固體。為了改進分離,在TFA-介導的脫保護反應后改進了方法中的兩個操作。第一個為低溫相分離;第二個為頭孢洛扎TFA的沉淀。產物萃取–低溫相分離可以用于分離頭孢洛扎TFA的低溫相分離由以下構成:用二氯甲烷稀釋脫保護反應混合物和冷卻批料至-40至-25℃。該方案產生雙相混合物,其中分離下層的頭孢洛扎富集相(基于溶劑密度;使用質量流量計測量)并在方法中繼續進行。當將該方案應用于鈀介導的偶聯后的工藝流時,由于頭孢洛扎TFA分布到雙相混合物的上層中,因此產生了顯著的產物損失(平均10-20%)。為了防止發生該問題,開發了如下所述新方案。在完成TFA-介導的脫保護反應之后,將批料用8-14體積的甲苯(目標10體積)稀釋。然后將所得的雙相混合物冷卻至約-25至約-5℃范圍內的溫度,得到容易觀察到的具有相分離的混合物(即,不需要流量計來監測分離)。下層的頭孢洛扎富集相容易從上層分離,產生工藝流的純度的顯著提升。此外,在該操作中發生非常少的產率損失,僅<1%的頭孢洛扎TFA殘留在被棄去的上層中。除了甲苯之外,其它非極性溶劑是可接受的,諸如烴溶劑(例如戊烷類、己烷類、庚烷類)、其它芳族溶劑(例如二甲苯、異丙基苯)及其一種或多種的混合物。分離–沉淀頭孢洛扎TFA當試圖在鈀-介導的偶聯后使用已知方法沉淀固體頭孢洛扎TFA時,典型的結果是形成凝膠樣的物質,其不能以可使用的方式過濾或分離。這可能是因為頭孢洛扎TFA在分離條件下傾向于團聚或在分離過程中頭孢洛扎TFA溶液的差的溶解度。這些問題以兩種方式解決。第一,在沉淀事件之前將額外的乙腈(溶劑)添加到批料中。這是違反常理的,因為加入額外的溶劑將通常不改善沉淀,這是由于額外的溶劑將會溶解待分離的材料。然而,在該情況下,額外的乙腈在沉淀中會穩定頭孢洛扎TFA溶液,有助于有序的和受控的沉淀。不希望受理論所限制,據信乙腈的添加會提供呈現出對于沉淀而言在動力學上有利的條件的溶劑混合物。優選地,在沉淀期間存在的約2至約6體積的乙腈確保了可過濾的固體的形成。由于溶劑的體積通常是相對于限制性試劑(在該情況下,如表1中所示的TATD-CLE)來確定,因此對于每100g限制性試劑而言,優選將約200至約600mL乙腈用于該目的。分離的第二個方案變化是將分離溫度降低至約0至約15℃。降低溶液溫度之后,添加反溶劑,諸如烴或醚類反溶劑,例如甲基叔丁基醚,以沉淀期望的產物。溫度的降低連同上面所述的額外的乙腈共同提供了高度可過濾的固體的受控的和順序的沉淀,并且不形成凝膠樣材料,由此在過濾后最大化了頭孢洛扎TFA的分離收率。7.5.3注釋該方法的總收率為78%(46.4g活性物,總共為85.9g),分離為淺褐色至黃色的固體。總純度為90.0%AUC(“曲線下面積”測量,參見例如圖5中的HPLC跡線圖),重量測定為52.0%。產物的鑒別可以通過與諸如圖6中所示的頭孢洛扎TFA的參比NMR譜圖比較來確認。所有摩爾當量和體積(mL/g)是相對于TATD-CLE的活性物的量。所有反應在氮氣環境下進行。頭孢洛扎TFA的摩爾收率為78%。計算頭孢洛扎TFA的分子量(不含抗衡離子)。產物的貯存和穩定性:在-20℃在透明玻璃容器中貯存材料。在這些條件下沒有觀察到降解,如通過HPLC分析確定的那樣。清洗操作:通常先用水,然后用丙酮清洗反應器,隨后煮沸丙酮并干燥。然后通過視覺檢查發現反應器已干凈。7.5.4分析方法和色譜圖表3.原位監測表4.方法表征的分析測試將表4的抽樣方案用于方法表征。這些分析測試并不是在方法驗證和隨后的商業生產過程中所需的,而是用于研究和/或額外的方法表征。7.6實施例6:制備頭孢他啶下述實驗方案(化合物1taz→化合物3taz以及化合物3taz→化合物5taz)展示了使用上述鈀催化技術制備抗生素頭孢他啶的一個途徑。關鍵的轉化是化合物3taz→化合物5taz,其中化合物5taz為含有常規保護基的頭孢他啶。化合物5taz的標準化學脫保護提供頭孢他啶。7.6.1制備化合物3taz在0℃將1taz(3.98g,12.08mmol)在二甲基乙酰胺(26.7mL)中的溶液用甲磺酰氯(1.87mL,24.16mmol)處理。添加碳酸鉀(1.67g,12.08mmol)并將反應混合物在0℃攪拌。2h后,將EtOAc加入至反應混合物中,隨后加入2.4%HCl的水溶液。將水層去除并棄去,并將剩余的有機層用10%(w/v)NaCl溶液洗滌。將水層去除并棄去,并在0℃將有機層緩慢添加至2(ACLE·HCl,4.45g,10.98mmol)在H2O(13.4mL)和EtOAc(13.4mL)中的溶液中。使用三乙胺(3.83mL)在EtOAc(5.78mL)中的溶液維持pH在3.2-3.8之間。一旦添加完成,將反應混合物在0℃攪拌30mins直到通過HPLC分析指示反應完成。將固體NaCl(0.445g)添加至反應混合物中,將其攪拌20分鐘。然后將反應混合物過濾,并將有機層分離并用20%(w/v)NaCl洗滌。將有機層干燥(NaSO4),濃縮并將殘余物通過SiO2上的色譜法(7:1EtOAc/己烷)純化以得到作為白色固體的3taz。1HNMR(CDCl3,400MHz)δ7.95(s,1H),7.35(d,2H,J=8.8Hz),6.94(s,1H),6.90(d,2H,J=8.8Hz),6.32(s,2H),5.99(d,1H,J=5.2Hz),5.27(d,1H,J=11.6Hz),5.20(d,1H,J=11.6Hz),5.05(d,1H,J=4.8Hz),4.55(d,1H,J=12.0Hz),4.45(d,1H,J=12.0Hz),3.81(s,3H),3.65(d,1H,J=18.0Hz),3.48(d,1H,J=18.0Hz),1.63(s,3H),1.60(s,3H),1.42(s,9H);HRMSESIm/zC29H35ClN5O8S2計算值680.1537[M+H]+,實測值680.1577。化合物3taz的HPLC色譜圖示于圖7中。7.6.2制備化合物5taz在30℃向3taz(0.250g,0.371mmol)在THF(2.15mL)中的溶液中添加KTFA(三氟乙酸鉀,0.085g,0.556mmol)、吡啶(0.033mL,0.408mmol)、4-phos(0.013g,0.030mmol)和Pd2dba3(0.003g,0.004mmol)。15min后,HPLC分析指示99%轉化(圖7和8)。將反應混合物過濾以得到5taz的黃色溶液。1HNMR(CD3OD,400MHz)δ9.01(d,2H,J=6.0Hz),8.61(t,1H,J=7.8Hz),8.10(dd,2H,J=6.8,7.6Hz),7.34(d,2H,J=8.8Hz),6.89(d,2H,J=8.8Hz),6.85(s,1H),5.98(d,1H,J=4.8Hz),5.74(d,1H,J=14.8Hz),5.40(d,1H,J=14.8Hz),5.26(dd,2H,J=12,21.2Hz),5.25(d,1H,J=5.2Hz),3.79(s,3H),3.70(d,1H,J=18.4Hz),3.40(d,1H,J=18.4Hz),1.54(s,3H),1.53(s,3H),1.45(s,9H);13CNMR(CD3OD,101MHz)δ175.20,171.41,165.60,165.21,162.93,161.61,149.69,147.64,146.00,142.92,131.90,131.11,129.65,127.89,121.76,114.98,111.65,84.24,83.20,69.84,62.21,60.74,59.29,55.76,28.22,27.40,24.59,24.49;HRMSESIm/zC34H39N6O8S2計算值723.2265[M]+,實測值723.2233。由化合物3taz形成化合物5taz的反應的HPLC色譜圖示于圖8中。7.7實施例7:去除鈀的水性后處理在完成了上述鈀-介導的偶聯反應后,使用水性后處理來降低在產物流中剩余的Pd水平。其實施例包括:1.用7體積的乙酸乙酯稀釋反應混合物。用7體積的5%EDTA水溶液洗滌所得漿料30分鐘。有機層的Pd含量從461ppm降低至370ppm。2.用7體積的乙酸乙酯稀釋反應混合物。用7體積的5%檸檬酸水溶液洗滌所得漿料30分鐘。有機層的Pd含量從461ppm降低至208ppm。3.用7體積的乙酸乙酯稀釋反應混合物。用7體積的1N三氟乙酸水溶液洗滌所得漿料30分鐘。有機層的Pd含量從700ppm降低至237ppm。4.用7體積的乙酸乙酯稀釋反應混合物。用7體積的5%氯化鈉水溶液洗滌所得漿料30分鐘。用13重量%DarcoG-60活性炭處理有機層30分鐘。有機層的Pd含量從461ppm降低至269ppm。5.將具有約100ppm鈀水平的頭孢洛扎TFA(100.0g游離堿當量,1當量)一次性加入至2升水中并將批料攪拌30分鐘,得到具有約1.6的pH的棕色溶液。隨后,將5%氫氧化銨溶液(165-170mL)添加到批料中直到溶液的pH為6.0至7.0,目標pH為約6.5。在達到目標pH后,加入5%二乙基二硫代氨基甲酸鈉的水溶液。通過將二乙基二硫代氨基甲酸鈉三水合物(0.5g,0.5%重量/重量,與頭孢洛扎游離堿相比)溶解在水(95.0g,9.5mL)中制備溶液。然后將批料攪拌30至60分鐘(目標時間為45分鐘),并經0.4微米過濾器過濾以去除含有鈀的固體。之后,將批料用15%鹽酸(105mL)處理直到溶液的pH為1.2-2.0(目標pH為1.5)。然后將批料攪拌20至40min(目標為30min),并使用0.2微米過濾器過濾以去除含有鈀的固體。然后如前所述將得到的包含頭孢洛扎和0.001-10ppm的殘余鈀水平的溶液轉化成具有相同的殘余鈀水平的頭孢洛扎硫酸鹽。8.等同方案和通過引用并入本申請中引用的所有出版物、專利、專利申請和其它文件通過引用以其整體并入本申請(為了所有目的),其程度與如同每篇單獨的出版物、專利、專利申請或其它文件被單獨地指示為通過引用并入(為了所有目的)一樣。盡管已經舉例說明和描述了多個具體實施方案,但是應理解可以做出多種改變而不偏離本發明的主旨和范圍。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1