大麗輪枝菌VdKu80基因敲除突變體菌株,其構建方法及其應用與流程
本發明涉及大麗輪枝菌突變體菌株及其應用,特別涉及敲除VdKu80基因的大麗輪枝菌突變體菌株,還涉及以所述大麗輪枝菌突變體菌株為受體菌株進行基因定點敲除的應用,屬于真菌分子生物學領域。
背景技術:
大麗輪枝菌(Verticillium dahliae Kleb.)是一種重要的植物病原真菌,屬于子囊菌門、糞殼菌科、輪枝菌屬。它在全世界范圍內有200種以上的寄主,包括喬木、灌木、經濟作物和觀賞性花卉等。由其引起的病害會導致植物枯萎死亡,因此造成了巨大的經濟損失。另外,大麗輪枝菌也是導致北京香山黃櫨枯萎病的病原真菌,每年造成大量的黃櫨枯萎死亡,嚴重的影響了香山紅葉的生態價值和景觀價值。對于由大麗輪枝菌引起的枯萎病,目前主要防治措施包括a.物理方法:加強營林措施、施肥灌溉增強樹勢、及時清理病害木殘體防止發生再次侵染;b.化學方法:使用各種農藥灌根、直接注射至受害植物以及使用化學農藥熏蒸處理土壤;c.生物方法:篩選對大麗輪枝菌具有拮抗作用的真菌(木霉)和細菌(枯草芽孢桿菌)、選育抗病植物。但是目前使用的這些方法防治效果并不理想。導致其難以防治主要有兩個原因:1、大麗輪枝菌主要危害寄主的維管束,一般的化學農藥以及拮抗菌無法發揮功能,因此效果大打折扣;2、大麗輪枝菌在其病害循環的后期會形成黑色素化的休眠結構“微菌核”,該結構由膨脹分隔的菌絲聚集而成,對于不良的外界環境具有極強的抗逆性,并且能夠在土壤內存活數年以上,一旦遇到合適的寄主,微菌核就會萌發形成菌絲,作為初侵染源,繼續對植物進行危害。
隨著科學技術的發展,人們開始大量研究大麗輪枝菌致病的分子機制以及大麗輪枝菌微菌核形成的分子機制,從而為大麗輪枝菌的防治提供理論依據。這方面的工作在農業有害病原真菌稻瘟菌(Magnaporthe oryzae)中取到了較大進展,大量參與附著胞形成及侵染過程的相關基因功能被研究,明確了它們在稻瘟菌侵染及致病的過程中發揮的功能并解析了它們之間的調控網絡,為稻瘟菌的防治提供了巨大的價值。因此,通過基因功能研究揭示大麗輪枝菌致病分子機制和微菌核形成分子機制,對于病害的防治意義重大。
對于大麗輪枝菌的基因功能研究,近十年來取得到了巨大的進展,尤其是在大麗輪枝菌全基因序列公布之后。大麗輪枝菌共有10535個編碼基因,而且隨著研究的進一步深入,為了更好的解析大麗輪枝菌的致病及微菌核形成的分子機制,開展高通量的基因功能研究是十分必要的。但是目前國內外已經研究功能的基因數量還非常有限,造成這一現象的一個重要原因是其同源重組率低,導致基因敲除效率不高。雖然近年來,研究者通過改變載體構建方法從double joint PCR到Split-marker、改變轉化方法從PEG介導的原生質體轉化到農桿菌介導的轉化等都有效的提高了基因敲除效率,但是目前基因敲除效率依然是限制大麗輪枝菌基因功能研究的主要因素之一。因此,提高大麗輪枝菌基因敲除效率對加快大麗輪枝菌基因功能研究的進程具有重要的意義。
在真核生物中,細胞會通過同源重組(homologous recombination)和非同源末端連接(non-homologous end joining)等方式修復受到破壞的雙鏈DNA,通常非同源末端連接方式是真核生物中主要的修復方式,它能抑制外源DNA在細胞內發生同源重組事件,因此該方式能有效的降低同源重組率,從而導致基因敲除效率降低。參與非同源末端連接修復方式的基因主要包括Ku70、Ku80、XRCC4、DNAPKcs和LIG4等,其中Ku70和Ku80組成的Ku異質二聚體負責識別受損的雙鏈DNA,因此它們在非同源末端連接修復途徑中發揮著重要的作用。大量的研究表明敲除Ku70或Ku80基因能夠大大的提高真菌的基因敲除率,這一現象在米曲霉(Aspergillus oryzae)、稻瘟菌(M.oryzae)、灰葡萄孢(Botrytis cinerea)、粗糙脈胞菌(Neurospora crassa)、黃青霉(Penicillium chrysogenum)等很多真菌中得到了驗證。已有研究顯示,敲除大麗輪枝菌Ku70基因也能夠顯著提高基因敲除率,從原有的0.5%提高到了20%以上,因此這一機制在大麗輪枝菌中也同樣適用。但是對于敲除Ku80基因能否顯著提高其基因敲除率并沒有研究。
故本發明旨在獲得具有高效基因敲除效率的大麗輪枝菌菌株。本發明通過PEG介導的原生質體轉化方法,成功敲除大麗輪枝菌VdKu80基因。結果表明△VdKu80突變體菌株其基因敲除率較野生型菌株得到了顯著的提高,并且大麗輪枝菌△VdKu80突變體菌株在菌絲生長、產孢、脅迫響應、微菌核形成以及致病性方面與野生型菌株無顯著差異。因此本發明獲得的大麗輪枝菌△VdKu80突變體菌株可以用于后續的基因功能研究。該突變體菌株的使用將大大提高大麗輪枝菌基因功能研究效率。
技術實現要素:
本發明的目的是針對現有大麗輪枝菌存在基因同源重組率低,基因敲除率低的技術問題,提供一株大麗輪枝菌(Verticillium dahliae)XS11的VdKu80基因敲除突變體菌株、其構建方法以及應用。本發明方法構建的VdKu80基因敲除突變體菌株在敲除VdKu80基因后,其基因敲除效率顯著提高,而且敲除VdKu80基因并不影響突變體菌株的菌絲生長、產孢、脅迫響應、微菌核形成和致病性。因此,敲除VdKu80基因的大麗輪枝菌突變體菌株能夠作為受體菌株對大麗輪枝菌其他基因進行敲除實驗。
為實現本發明的目的,本發明一方面提供一株大麗輪枝菌(Verticillium dahliae)XS11的VdKu80基因敲除突變體菌株VdXS11-△VdKu80,其微生物保藏號為CGMCC No.12873。
其中,所述VdKu80基因被潮霉素抗性基因所替代。
本發明另一方面提供一種大麗輪枝菌(Verticillium dahliae)的VdKu80基因敲除突變體菌株VdXS11-△VdKu80的構建方法,包括如下步驟:1)利用Split-marker方法構建大麗輪枝菌VdKu80基因敲除載體;2)利用PEG介導的方法,將VdKu80基因敲除載體導入野生型大麗輪枝菌的原生質體內,獲得大麗輪枝菌VdKu80基因敲除的轉化子;3)利用PCR方法對大麗輪枝菌VdKu80基因敲除轉化子進行篩選。
其中,步驟1)中所述大麗輪枝菌VdKu80基因敲除載體的構建方法,包括如下步驟:
1A)以大麗輪枝菌基因組DNA為模板,分別以Ku80-5F/Ku80-5R、Ku80-3F/Ku80-3R為特異性引物進行PCR擴增,獲得大麗輪枝菌VdKu80基因上游片段VdKu80 5F、下游片段VdKu80 3F;
1B)以質粒gGFP為模板,以Hyg-For/Hyg-Rev為特異性引物進行PCR擴增,獲得潮霉素抗性基因片段Hygromycin片段;
1C)將VdKu80 5F、VdKu80 3F片段、Hygromycin片段分別與pMDTM19-T Vector質粒進行連接反應,獲得VdKu80 5F重組質粒、VdKu80 3F重組質粒和Hygromycin重組質粒;
1D)以VdKu80 5F重組質粒為模板,以Ku80-5F/Ku80-5RO為引物,進行PCR擴增得到VdKu80 5FO片段;以VdKu80 3F重組質粒為模版,以Ku80-3FO/Ku80-3R為引物,進行PCR擴增得到VdKu80 3FO片段;以Hygromycin重組質粒為模版,以M13F/M13R為引物,進行PCR擴增得到HygromycinO片段;
1E)以VdKu80 5FO片段和HygromycinO片段為模板,以Ku80-5F/HY-R為引物,進行融合PCR擴增,獲得大麗輪枝菌VdKu80基因敲除載體1:VdKu80 5F+2/3Hyg片段;以VdKu80 3FO片段和HygromycinO片段為模板,以YG-F/Ku80-3R為引物,進行融合PCR擴增,獲得大麗輪枝菌Ku80基因敲除載體2:2/3Hyg+VdKu80 3F片段。
特別是,步驟1D)中VdKu80 5FO片段在3’端含有一段與M13F相同的DNA序列;VdKu80 3FO片段在5’端含有一段與M13R相同的DNA序列;HygromycinO片段的5’和3’分別含有M13F和M13R的DNA序列。
其中,步驟2)中所述野生型大麗輪枝菌菌株為大麗輪枝菌XS11菌株。
特別是,步驟2)中所述原生質體轉化包括如下步驟:
2A)將野生型大麗輪枝菌分生孢子置于液體YEPD培養基中培養,得到大麗輪枝菌新鮮菌絲。
2B)利用Lyzing和Driselase酶,對大麗輪枝菌新鮮菌絲進行酶解,獲得原生質體;
2C)將大麗輪枝菌VdKu80基因敲除載體VdKu80 5F+2/3Hyg片段和2/3Hyg+VdKu80 3F片段與大麗輪枝菌原生質體混合后,在一定濃度的PEG條件下導入大麗輪枝菌原生質體內,獲得大麗輪枝菌VdKu80基因敲除的轉化子。
其中,步驟3)中所述PCR擴增方法篩選大麗輪枝菌VdKu80基因敲除轉化子包括如下步驟:
3A)使用CTAB法提取大麗輪枝菌VdKu80基因敲除轉化子的DNA;
3B)以步驟3A)提取的DNA為模板,以Ku80-IF/Ku80-IR為引物,對所有轉化子進行PCR擴增;
3C)對PCR擴增產物進行凝膠電泳檢測,PCR擴增產物中未能擴增出Ku80基因片段(102bp大小)條帶的轉化子篩選為大麗輪枝菌VdKu80基因敲除突變體。
特別是,還包括步驟4),采用Southern blot方法對大麗輪枝菌VdKu80基因敲除轉化子進行分析、鑒定。
其中,所述大麗輪枝菌VdKu80基因敲除轉化子的分析、鑒定包括如下步驟:
4A)制備探針
以VdKu80 3F重組質粒為模板,以Ku80-3F/Ku80-P1為引物,進行PCR擴增獲得探針片段;接著使用DIG High Prime DNA Labling and Detection Starter Kit I(Roche)試劑盒中的DIG-High Prime并按照試劑盒說明書對探針片段進行標記,制得地高辛標記探針;
4B)雜交處理
使用CTAB法提取大麗輪枝菌VdKu80基因敲除轉化子的DNA;接著用KpnI限制性內切酶對DNA進行酶切處理;然后將酶切后的DNA片段經瓊脂糖凝膠電泳分離后轉移至N+的硝酸纖維素膜上;再利用地高辛(DIG)標記探針進行雜交。
特別是,還包括對雜交后的硝酸纖維素膜按照DIG High Prime DNA Labling and Detection Starter Kit I(Roche)試劑盒的要求進行洗膜,顯色。
本發明又一方面提供一種大麗輪枝菌VdKu80基因敲除突變體菌株VdXS11-△VdKu80-9作為受體菌株在提高基因敲除效率中的用途。
本發明與現有技術相比,具有以下優點:
本發明通過PEG介導的原生質體轉化方法,成功敲除大麗輪枝菌VdKu80基因,獲得敲除Ku80基因的突變體菌株VdXS11-△VdKu80-9,該突變體菌株的基因敲除率較野生型菌株得到了顯著的提高,基因敲除率提高20%以上;而且本發明的大麗輪枝菌△VdKu80突變體菌株在菌絲生長、產孢、脅迫響應、微菌核形成以及致病性方面與野生型菌株無顯著差異。因此本發明獲得的大麗輪枝菌△VdKu80突變體菌株可以用于后續的基因功能研究,并且能大大提高大麗輪枝菌基因功能研究效率。
附圖說明
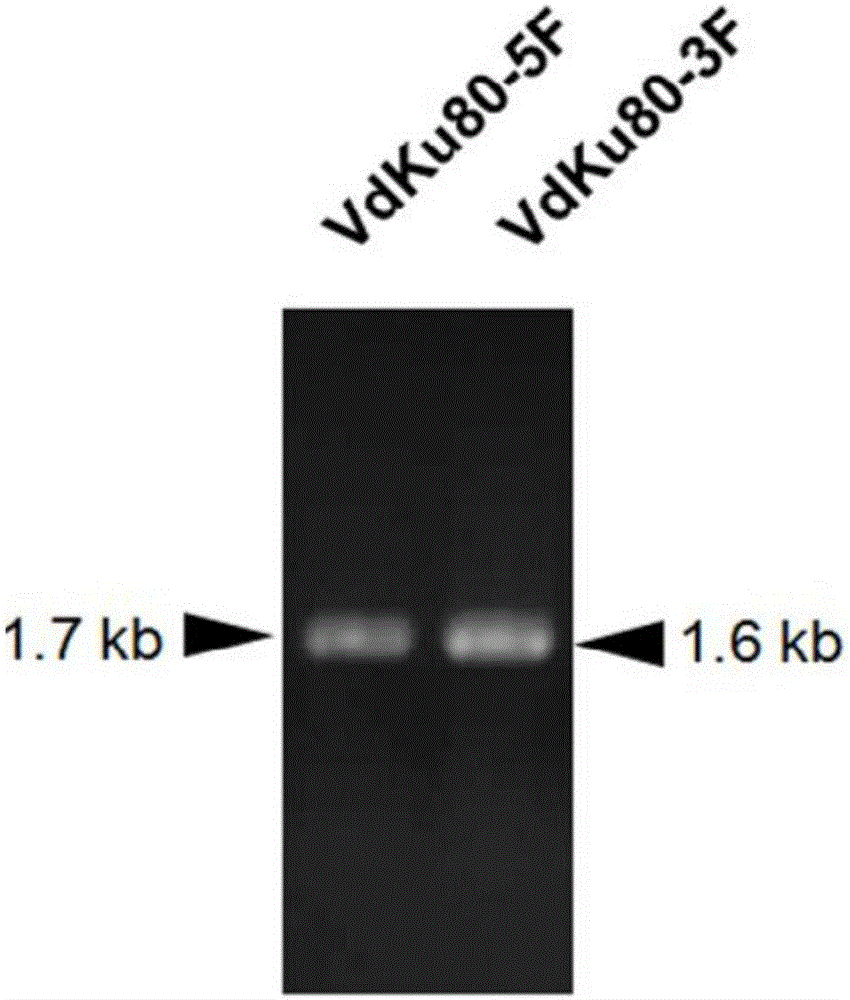
圖1A是VdKu80基因上游、下游片段的電泳檢測圖;
圖1B是VdKu80基因上游、下游片段分別融合2/3Hygromycin片段的電泳檢測圖;
圖2A是利用Ku80-IF/Ku80-IR引物進行PCR驗證VdKu80基因敲除突變體的電泳檢測圖;
圖2B是利用Southern blot鑒定VdKu80基因敲除突變體的檢測圖;其中:
WT表示大麗輪枝菌野生型;△VdKu80_1、△VdKu80_4、△VdKu80_6、△VdKu80_9表示成功敲除了VdKu80基因的敲除突變體;VdKu80_5表示構建的VdKu80敲除片段異位整合到基因組上其他位置上了。用于Southern blot分析的野生型和突變體基因組均使用KpnI限制性內切酶進行酶切。
圖3A是大麗輪枝菌野生型菌株和△VdKu80突變體菌株在培養基PDA、CM、CM+100μg/ml Cong Red、CM+1M NaCl上生長的菌落直徑統計結果圖;
圖3B是大麗輪枝菌野生型菌株和△VdKu80突變體菌株在培養基PDA、CM、CM+100μg/ml Cong Red、CM+1M NaCl上生長菌落形態圖;
圖3C是大麗輪枝菌野生型菌株和△VdKu80突變體菌株在培養基PDA上生長10天后的產孢量統計結果圖。
圖4是大麗輪枝菌野生型菌株和△VdKu80突變體菌株在BM培養基上培養5天后,微菌核形成的顯微鏡觀察;
圖5是大麗輪枝菌野生型菌株和△VdKu80突變體菌株對煙草幼苗致病性測定,其中:CK蒸餾水對照組,將煙草幼苗的根部浸泡在含106孢子/ml的野生型菌株和△VdKu80突變體菌株的孢子懸浮液中10分鐘,對照使用蒸餾水。圖片拍攝于接種后25天。
具體實施方式
下面結合具體實施例來進一步描述本發明,本發明的優點和特點將會隨著描述而更為清楚。但這些實施例僅是范例性的,并不對本發明的范圍構成任何限制。本領域技術人員應該理解的是,在不偏離本發明的精神和范圍下可以對本發明技術方案的細節和形式進行修改或替換,但這些修改和替換均落入本發明的保護范圍內。
下述實施例中的方法,無特別說明,均為常規方法。PCR反應中所用的DNA聚合酶、dNTP,提取基因組的試劑盒和PCR產物純化的試劑盒等分子生物學常規試劑均購自寶生物工程(大連)有限公司和天根生化科技(北京)有限公司,相應的分子生物學操作按照商品試劑說明書和《分子克隆》(第三版)進行。
本發明所涉及的術語除另外定義之外,所使用的技術及術語均與本發明所屬領域普通技術人員通常所了解的具有相同含義。
實施例1材料、試劑、培養基配制
1、野生型菌株和質粒
大麗輪枝菌XS11作為本發明使用的野生型菌株,采集自香山地區枯萎的黃櫨病部組織。
質粒gGFP,用于擴增潮霉素基因片段,購自于美國堪薩斯州的真菌遺傳儲存中心(Fungal Genetics Stock Center)。
質粒pSilent-Dual1,用于擴增遺傳霉素基因片段,購自于美國堪薩斯州的真菌遺傳儲存中心(Fungal Genetics Stock Center)。
質粒pMDTM19-T Vector,用于基因克隆,購自于寶生物工程(大連)有限公司。
2、培養基
2-1)PDA固體培養基
200g土豆,20g D-glucose(D-葡萄糖),15g瓊脂粉,無菌水定容至1L,置于121℃,滅菌30min。
2-2)CM固體培養基
50ml 20×nitrate salts,1ml 1000x Trace,10g D-glucose,2g peptone(細菌蛋白胨),1g yeast extract(酵母浸膏),1g casamino acids(酸水解酪素),1ml vitamin solution,用NaOH調pH至6.5,無菌水定容至1L,瓊脂粉15g/L,置于121℃,滅菌30min。
2-3)脅迫培養基1(含有100μg/ml Cong Red(剛果紅)的CM固體培養基):
使用前每1L CM固體培養基中加入Cong Red 10mg(即Cong Red的濃度為100μg/ml)。
2-4)脅迫培養基2(含1M的NaCl的CM固體培養基)
使用前每1L CM固體培養基中加入58.5g的NaCl(即NaCl的濃度為1M)。
2-5)微菌核生長培養基(BM固體培養基)
10g D-glucose,0.2g sodium nitrate,0.52g KCl,0.52g MgSO4·7H2O,1.52g KH2PO4,3μM thiamine HCl,0.1μM biotin,15g瓊脂粉,無菌水定容至1L,置于121℃,滅菌30min。
2-6)Hygromycin抗性PDA固體培養基
使用前每1L PDA固體培養基中加入25mg潮霉素。
2-7)YEPD培養基,用于大麗輪枝菌菌絲培養
實施例2構建基因敲除載體
采用Split-marker方法對大麗輪枝菌VdKu80基因敲除載體進行構建。
1、擴增大麗輪枝菌VdKu80基因(VdKu80基因)上、下游片段
1A、根據大麗輪枝菌(Verticillium dahliae Kleb.)野生株XS11基因組VdKu80編碼基因的序列,設計2對PCR擴增特異性引物(引物名稱、序列如表1所示);其中:Ku80-5F/Ku80-5R為VdKu80基因上游片段引物;Ku80-3F/Ku80-3R為VdKu80基因下游片段引物;
1B、以大麗輪枝菌基因組(完整的基因序列參見http://genome.jgi.doe.gov/Verda1/Verda1.home.html)為模板,分別以Ku80-5F/Ku80-5R;Ku80-3F/Ku80-3R為引物進行PCR擴增,得到1.7kb的VdKu80基因上游片段(VdKu80 5F,SEQ ID NO.36);1.6kb的VdKu80基因下游片段(VdKu80 3F,SEQ ID NO.37);PCR擴增產物的1%瓊脂糖凝膠電泳檢測結果如圖1A所示。
PCR反應體系:
PCR反應條件:94℃預變性5min,94℃30s,55℃30s,72℃2min,30個循環,最后72℃延伸10min,以雙蒸水為陰性對照。
2、擴增潮霉素抗性基因片段(Hygromycin片段)
以質粒gGFP為模板,以Hyg-For/Hyg-Rev為引物(引物名稱、序列如表1所示),進行PCR擴增,獲得1.5kb的Hygromycin片段(即潮霉素抗性基因片段),包括啟動子、開放式閱讀框和終止子,委托英濰捷基(上海)貿易有限公司進行測序,SEQ ID NO.38。
3、制備重組質粒
3A、將VdKu80 5F、VdKu80 3F片段和Hygromycin片段分別與pMDTM19-T Vector質粒進行連接反應(按照pMDTM19-T Vector clone kit試劑盒說明進行),其中反應體系和條件如下:
目標片段 2μl
pMDTM19-T Vector 0.5μl
Solution I 2.5μl
37℃連接3h
3B、使用DH5α大腸桿菌感受態進行重組質粒的大腸桿菌轉化,并分別使用M13F/M13R引物對菌落進行陽性克隆篩選、將能夠分別擴增出VdKu80 5F、VdKu80 3F和Hygromycin片段目的條帶的菌落進行搖菌,并利用天根生化科技(北京)有限公司的質粒提取試劑盒提取質粒,獲得VdKu80 5F重組質粒、VdKu80 3F重組質粒、Hygromycin重組質粒。
4、含有重疊列片段的PCR擴增
以VdKu80 5F重組質粒為模版,以Ku80-5F/Ku80-5RO為引物,進行PCR擴增得到VdKu80 5F重疊擴增片段(即VdKu80 5FO片段);以VdKu80 3F重組質粒為模版,以Ku80-3FO/Ku80-3R為引物,進行PCR擴增得到VdKu80 3F重疊擴增片段(即VdKu80 3FO片段);以Hygromycin重組質粒為模版,以M13F/M13R為引物,進行PCR擴增得到Hygromycin重疊擴增片段(即HygromycinO片段);其中VdKu80 5FO片段在3’端含有一段與M13F相同的DNA序列;VdKu80 3FO片段在5’端含有一段與M13R相同的DNA序列;HygromycinO片段的5’和3’分別含有M13F和M13R的DNA序列;引物序列如表1所示。
PCR反應體系:
PCR反應條件:94℃預變性5min,94℃30s,55℃30s,72℃2min,30個循環,最后72℃延伸10min,以雙蒸水為陰性對照。
5、融合PCR
以VdKu80 5FO片段和HygromycinO片段為模板,以Ku80-5F/HY-R為引物(見表1),利用融合PCR方法進行PCR擴增,獲得VdKu80基因敲除載體片段1(即VdKu80 5F+2/3Hyg片段);以VdKu80 3FO片段和HygromycinO片段為模板,以YG-F/Ku80-3R為引物(見表1)進行PCR擴增,即通過融合PCR的方法擴增獲得VdKu80基因敲除載體片段2(即2/3Hyg+VdKu80 3F片段);融合PCR擴增產物經1%瓊脂糖凝膠電泳檢測獲得的VdKu80敲除載體片段1、2的大小分別為2.7kb、2.5kb。電泳檢測結果如圖1B。
融合PCR反應體系:
PCR反應條件:94℃預變性5min,94℃30s,55℃45s,72℃3min,30個循環,最后72℃延伸10min,以雙蒸水為陰性對照。
實施例3PEG介導的原生質體轉化
將VdKu80敲除載體VdKu80 5F+2/3Hyg和2/3Hyg+VdKu80 3F兩個片段使用PEG介導的遺傳轉化方法導入大麗輪枝菌野生型原生質體內。具體轉化方法參考(Goswami,RS.Targeted gene replacement in fungi using a split-marker approach.Methods Mol Biol,2011,835:255-269)的方法進行,具體操作如下:
1、將適量的野生型大麗輪枝菌菌株XS11的分生孢子置于液體的YEPD培養基中,于25℃,150rpm搖培一定時間獲得新鮮的大麗輪枝菌菌絲
2、利用適量比例的Lyzing和Driselase酶,對大麗輪枝菌新鮮菌絲進行酶解,獲得足量的原生質體。
3、將構建好的基因敲除載體VdKu80 5F+2/3Hyg和2/3Hyg+VdKu80 3F片段與大麗輪枝菌原生質體充分混合后,在一定濃度的PEG條件下導入大麗輪枝菌原生質體內。
4、原生質體再生培養
將一定量的TB3液體培養基加入步驟3中的大麗輪枝菌原生質體懸浮液中,于25℃,90rpm進行原生質體再生培養,促進原生質體細胞壁的再生,將細胞壁再生后的菌體置于含25μg/ml潮霉素的TB3固體培養基中生長,共獲得13個VdKu80基因敲除的候選轉化子。
實施例4基因敲除突變體篩選、鑒定
1、VdKu80基因敲除轉化子單胞純化
將VdKu80基因敲除轉化子轉接至含潮霉素的PDA固體培養基(即Hygromycin抗性PDA固體培養基,其中潮霉素濃度為25μg/mL)上,于室溫下黑暗培養7天后用蒸餾水沖洗菌落,收集轉化子的分生孢子,并用10倍梯度稀釋法稀釋孢子懸液;
將少量的稀釋后的孢子懸浮液涂布到新的含Hygromycin的PDA固體培養基上生長,其中潮霉素濃度為25μg/mL,室溫下黑暗培養2-3天,待菌落可見,挑取單個菌落于無Hygromycin抗性的PDA平板(即不含潮霉素的PDA固體培養基)上生長,室溫下黑暗培養5-7天,用1ml蒸餾水沖洗菌落,收集單胞轉化子的分生孢子,并用15%甘油進行保存,置于-80℃待用。經初步篩選,挑取5個單胞轉化子(分別為VdKu80-1、VdKu80-4、VdKu80-5、VdKu80-6、VdKu80-9)作為VdKu80基因敲除的待選轉化子用于進一步分析驗證。
2、突變體的初步篩選
使用CTAB(十六烷基三甲基溴化銨,cetyltrimethylammonium Ammonium Bromide)的方法分別提取每個單胞轉化子、野生型菌株(WT)的基因組DNA,使用引物Ku80-IF/Ku80-IR(序列見表1)進行PCR擴增檢測。
PCR擴增產物的瓊脂糖凝膠電泳檢測結果如圖2A,野生型菌株和轉化子VdKu80-5能夠擴增出目的條帶(即102bp的VdKu80基因片段),而轉化子VdKu80-1、VdKu80-4、VdKu80-6、VdKu80-9未擴增出目的條帶。
轉化子中VdKu80基因被敲除,則PCR擴增為陰性結果,即PCR擴增產物中不能擴增出102bp大小的目的條帶(即VdKu80基因片段),如果仍能擴增出與野生型條帶大小(102bp)一樣的的產物,則說明VdKu80基因未被敲除。
圖2A的檢測結果顯示:大麗輪枝菌野生型菌株(WT)具有明顯的102bp大小的目的條帶;轉化子VdKu80-5的電泳圖中具有與野生型一樣大小的條帶;而轉化子VdKu80-1、VdKu80-4、VdKu80-6、VdKu80-9的PCR擴增產物中未能擴增出102bp大小的條帶,因此初步篩選出轉化子VdKu80-1、VdKu80-4、VdKu80-6、VdKu80-9為VdKu80基因敲除突變體命名為△VdKu80-1、△VdKu80-4、△VdKu80-6、△VdKu80-9。
3、Southern blot分析、鑒定
使用DIG High Prime DNA Labling and Detection Starter Kit I(Roche)試劑盒,并嚴格按照其說明進行操作,對VdKu80敲除突變體進行Southern blot分析、鑒定,具體如下:
3A、以VdKu80 3F重組質粒為模板,以Ku80-3F/Ku80-P1為引物(見表1),進行PCR擴增,然后以DIG-High Prime(DIG High Prime DNA Labling and Detection Starter Kit I(Roche)試劑盒)進行標記,獲得DIG(地高辛)標記探針;
3B、將大麗輪枝菌野生型菌株、單胞轉化子的基因組DNA各約30μg,分別用限制性內切酶KpnI酶(約300U)進行酶切處理;將酶切后的DNA片段經瓊脂糖凝膠電泳分析后轉移至N+的硝酸纖維素膜上;再利用地高辛(DIG)標記探針進行雜交;然后按照DIG High Prime DNA Labling and Detection Starter Kit I(Roche)試劑盒中的說明對雜交后的硝酸纖維素膜進行洗膜、顯色、拍照,結果如圖2B所示。
在Southern blot分析、鑒定試驗中,野生型經過Southern blot分析雜出一條5.2kb的條帶(含有Ku80基因片段);成功敲除VdKu80基因的突變體未檢測到該條帶,而是雜出一條3.5kb的條帶(含有潮霉素基因片段);異位突變體雜出了2條條帶,一條與野生型雜出的條帶大小一致,另一條與VdKu80敲除突變體雜出的條帶大小一致。
由圖2A、2B的檢測結果可知本發明成功獲得4株單拷貝敲除VdKu80基因的大麗輪枝菌突變體菌株,潮霉素抗性基因成功整合并替代了VdKu80基因,命名為VdXS11-△VdKu80-1、VdXS11-△VdKu80-4、VdXS11-△VdKu80-6、VdXS11-△VdKu80-9。本發明將大麗輪枝菌VdKu80敲除突變體菌株VdXS11-△VdKu80-9提交中國微生物菌種保藏管理委員會普通微生物中心進行保藏,其微生物保藏編號為:CGMCC No.12873,保藏于中國微生物菌株保藏管理委員會普通微生物中心;保藏地址:中國北京市朝陽區北辰西路1號院3號中國科學院微生物研究所;保藏日期:2016年8月17日。
實施例5基因敲除突變體表型特征分析
1、VdKu80敲除突變體生長速率、菌落形態、分生孢子產量測定。
1A、生長速率測定:
將新鮮生長的大麗輪枝菌VdKu80基因敲除突變體和野生型大麗輪枝菌菌株的菌絲塊(直徑0.5cm)分別接種于PDA、CM固體平板培養基中央,室溫條件下培養10天后,分別測量菌落直徑,測定結果如圖3A,作為衡量其生長速率的標準。
測定結果表明:大麗輪枝菌野生型菌株與敲除VdKu80基因的突變體菌株在PDA或者CM固體培養基上菌落直徑無顯著差異。表明敲除VdKu80基因不影響真菌的生長。
1B、菌落形態觀察:
將新鮮生長的大麗輪枝菌VdKu80基因敲除突變體和野生型大麗輪枝菌菌株的菌絲塊(直徑0.5cm)分別接種于PDA、CM固體平板培養基中央,室溫條件下培養,直接拍照觀察菌落形態,觀察結果如圖3B。
VdKu80敲除突變體菌株和野生型菌株不論在PDA培養基上還是在CM培養基上,菌落形態均無顯著差異。表明敲除VdKu80基因不影響真菌的菌落形態。
1C、分生孢子產量測定:
將新鮮生長的大麗輪枝菌VdKu80基因敲除突變體和野生型大麗輪枝菌菌株的菌絲塊(直徑0.5cm)分別接種于PDA固體平板培養基中央,室溫條件下培養10天,然后用2ml蒸餾水沖洗菌落,獲得分生孢子懸浮液,并用血球計數板對分生孢子數量進行統計,統計結果如圖3C。
由圖3C可知:本發明的VdKu80敲除突變體菌株和野生型菌株在分生孢子產量方面無顯著不同,均產生大量分生孢子,室溫條件下培養10天后的分生孢子產量為2.4-2.6×108個/ml,表明敲除大麗輪枝菌的VdKu80基因,并不影響菌株的生長、菌落形態和分生孢子產量。
2、脅迫響應
將新鮮生長的大麗輪枝菌VdKu80基因敲除突變體和野生型大麗輪枝菌菌株的菌絲塊(直徑0.5cm)分別接種至含有100μg/ml Cong Red(剛果紅)、1M/L的NaCl的CM固體平板培養基的中央,室溫條件下生長10天,測量大麗輪枝菌野生型和VdKu80敲除突變體菌株的菌落直徑,測定結果如圖3A所示;觀察菌落形態,觀察結果如圖3B所示。
測定結果表明:野生型菌株和VdKu80基因敲除突變體菌株在脅迫培養基上的菌絲生長較無脅迫的CM固體培養基,生長受到了顯著的抑制。尤其是在含1M/L NaCl的CM固體培養基。但是不論是在含100μg/mlCong Red的CM固體培養基還是在含1M/L NaCl的CM固體培養基,野生型菌株和VdKu80基因敲除突變體菌株菌落直徑和菌落形態均無顯著差異。表明,VdKu80敲除突變體菌株和野生型菌株對細胞壁脅迫劑和滲透壓力的抗性一致,敲除VdKu80基因并影響大麗輪枝菌對外界壓力的響應。
3、微菌核形成觀察
將1×106個孢子/ml的VdKu80敲除突變體和野生型菌株的孢子液均勻的涂布在BM固體培養基上,室溫條件下生長5天后,用顯微鏡觀察微菌核形成情況,顯微鏡觀察結果如圖4。
VdKu80基因敲除突變體菌株能夠形成與野生型菌株類似的黑色素化的微菌核,形成的微菌核無顯著差異,敲除VdKu80基因并不影響大麗輪枝菌微菌核的形成。
4、致病性實驗
將煙草幼苗的根分別浸泡在分生孢子為1×106個/ml的VdKu80敲除突變體、野生型菌株的孢子懸浮液中10min,然后將煙草重新種植到土壤內,至于溫室內生長20-30天后觀察煙草幼苗的生長狀況以及枯萎病癥;對照組(CK)除了采用蒸餾水浸泡煙草幼苗根之外,其余與分生孢子懸浮液浸泡相同。觀察結果如圖5所示。
經大麗輪枝菌VdKu80基因敲除突變體菌株與野生型菌株浸泡后的煙草幼苗生長狀況差,出現了明顯的枯萎病癥狀包括葉片黃萎、脫落;與對照組煙草幼苗相比,接種了麗輪枝菌VdKu80基因敲除突變體菌株與野生型菌株的煙草幼苗生長受到顯著的抑制;而且接種大麗輪枝菌VdKu80基因敲除突變體菌株的煙草幼苗與接種大麗輪枝菌野生型菌株的煙草幼苗表型無顯著差異。實驗表明,敲除大麗輪枝菌VdKu80基因并不影響大麗輪枝菌的致病性。
綜上所述,本發明獲得的大麗輪枝菌VdKu80敲除突變體菌株在形態上與野生型菌株無顯著差異,同時在大麗輪枝菌中敲除VdKu80并不影響菌株的生長、分生孢子的產量、脅迫響應能力、微菌核形成和致病性。本發明獲得的VdKu80敲除突變體菌株與野生型菌株在各方面表型上均無顯著差異,因此本發明獲得的VdKu80敲除突變體菌株可以作為后續基因功能研究的受體菌株,用于基因敲除。
由于VdKu80基因敲除突變體菌株中含有潮霉素抗性篩選基因,因此本實例中使用的是遺傳霉素(Geneticin,G418)抗性基因替換VDAG_00736基因、VDAG_07169基因。
試驗例1以VdKu80敲除突變體菌株VdXS11-△VdKu80-9作為受體菌株進行VDAG_00736基因敲除效率評估
采用Split-marker方法對大麗輪枝菌VdKu80敲除突變體菌株VdXS11-△VdKu80-9的VDAG_00736基因敲除載體進行構建。
1.1構建VDAG_00736基因敲除載體
按照實施例2構建VdKu80基因敲除載體的方法,構建了VDAG_00736融合遺傳霉素抗性的敲除片段VDAG_00736-5F+2/3G418和2/3G418+VDAG_00736-3F。
以大麗輪枝菌基因組DNA(完整的基因序列參見http://genome.jgi.doe.gov/Verda1/Verda1.home.html)為模板,分別以根據大麗輪枝菌VDAG_00736基因的上、下游DNA序列所設計2對PCR擴增特異性引物VDAG_00736-5F/VDAG_00736-5R;VDAG_00736-3F/VDAG_00736-3R為引物進行PCR擴增,獲得大麗輪枝菌VDAG_00736基因上游片段VDAG_00736 5F和VDAG_00736基因下游片段VDAG_00736 3F;
以質粒pSilent-Dual1為模板,以根據質粒pSilent-Dual1的DNA序列而設計1對遺傳霉素抗性基因PCR擴增的特異性引物Geneticin-For/Geneticin-Rev為引物進行PCR擴增,獲得Geneticin片段(即遺傳霉素抗性基因片段),包括啟動子、開放式閱讀框和終止子;
將VDAG_00736 5F、VDAG_00736 3F、Geneticin片段分別與pMDTM19-T Vector質粒進行連接反應,使用DH5α大腸桿菌感受態進行重組質粒的大腸桿菌轉化,并分別使用M13F/M13R引物對菌落進行陽性克隆篩選、利用天根生化科技(北京)有限公司的質粒提取試劑盒提取質粒,獲得重組質粒VDAG_00736 5F、VDAG_00736 3F、Geneticin;
以VDAG_00736 5F重組質粒為模版,以VDAG_00736-5F/VDAG_00736-5O為引物,進行PCR擴增得到VDAG_00736 5FO片段,該片段在3’端含有一段與M13F引物相同的DNA序列;以VDAG_00736 3F重組質粒為模版,以VDAG_00736-3O/VDAG_00736-3R為引物,進行PCR擴增得到VDAG_00736 3FO片段,該片段在5’端含有一段與M13R引物相同的DNA序列;以Geneticin重組質粒為模版,以M13F/M13R為引物,進行PCR擴增得到5’和3’分別含有M13F和M13R引物序列的GeneticinO片段;
以VDAG_00736 5FO片段和GeneticinO片段為模板,以VDAG_00736-5F/GE-R為引物,利用融合PCR方法進行PCR擴增,獲得VDAG_00736基因敲除載體片段A(即VDAG_00736-5F+2/3G418片段);以VDAG_00736 3FO片段和GeneticinO片段為模板,以EN-F/VDAG_00736-3R為引物,通過融合PCR的方法擴增獲得VDAG_00736基因敲除載體片段B(即2/3G418+VDAG_00736-3F片段);
1.2PEG介導的原生質體轉化
使用PEG介導的遺傳轉化方法,將構建的VDAG_00736敲除片段VDAG_00736-5F+2/3G418,2/3G418+VDAG_00736-3F導入本發明方法制備的VdXS11-△VdKu80-9敲除突變體菌株的原生質體內,獲得VDAG_00736基因敲除的待選轉化子,具體轉化方法參考(Goswami,R S.Targeted gene replacement in fungi using a split-marker approach.Methods Mol Biol,2011,835:255-269)的方法進行。
1.3VDAG_00736基因敲除突變體篩選和統計
將步驟2中獲得的獲得VDAG_00736基因敲除的待選轉化子轉接至含有50μg/ml Geneticin的PDA抗性平板培養基中,于室溫條件下黑暗培養7天,用1ml蒸餾水沖洗菌落,收集分生孢子;接著將少量分生孢子懸浮液均勻的涂布到含有50μg/ml Geneticin的PDA抗性平板培養基上進行單胞純化,挑取單胞轉化子;然后將純化后的轉化子保存并采用CTAB法提取基因組DNA;再接著以提取的轉化子的DNA為模板,以VDAG_00736-IF/VDAG_00736-IR為引物篩選VDAG_00736基因敲除突變體,并統計敲除突變體的比例(敲除突變體占所有篩選轉化子的比例),本試驗例中所使用的引物如表1所示,統計結果如表2。
試驗例2以VdKu80敲除突變體菌株VdXS11-△VdKu80-9作為受體菌株進行VDAG_07169基因敲除效率評估
采用Split-marker方法對大麗輪枝菌VdKu80敲除突變體菌株VdXS11-△VdKu80-9的VDAG_07169基因敲除載體進行構建。
1、構建VDAG_07169單基因敲除載體
按照實施例2構建VdKu80基因敲除載體的方法,構建了VDAG_07169融合遺傳霉素抗性的敲除片段VDAG_07169-5F+2/3G418和2/3G418+VDAG_07169-3F。
以大麗輪枝菌基因組DNA(完整的基因序列參見http://genome.jgi.doe.gov/Verda1/Verda1.home.html)為模板,分別以根據大麗輪枝菌VDAG_07169基因的上、下游DNA序列所設計2對PCR擴增特異性引物VDAG_07169-5F/VDAG_07169-5R、VDAG_07169-3F/VDAG_07169-3R為引物進行PCR擴增,獲得大麗輪枝菌VDAG_07169基因上游片段VDAG_07169 5F和VDAG_07169基因下游片段VDAG_07169 3F;
將VDAG_07169 5F和VDAG_07169 3F片段分別與pMDTM19-T Vector質粒進行連接反應,使用DH5α大腸桿菌感受態進行重組質粒的大腸桿菌轉化,并分別使用M13F/M13R引物對菌落進行陽性克隆篩選、利用天根生化科技(北京)有限公司的質粒提取試劑盒提取質粒,獲得VDAG_07169 5F重組質粒和VDAG_07169 3F重組質粒;
以VDAG_07169 5F重組質粒為模版,以VDAG_07169-5F/VDAG_07169-5O為引物,進行PCR擴增得VDAG_07169 5FO片段,該片段在3’端含有一段與M13F引物相同的DNA序列;以VDAG_07169 3F重組質粒為模版,以VDAG_07169-3O/VDAG_07169-3R為引物,進行PCR得VDAG_07169 3FO片段,該片段在5’端含有一段與M13R引物相同的DNA序列;
以VDAG_07169 5FO片段和試驗例1中獲得的GeneticinO片段為模板,以VDAG_07169-5F/GE-R為引物,利用融合PCR方法進行PCR擴增,獲得VDAG_07169基因敲除載體片段A(即VDAG_07169-5F+2/3G418片段);以VDAG_07169 3FO片段和試驗例1中獲得的GeneticinO片段為模板,以EN-F/VDAG_07169-3R為引物,通過融合PCR的方法擴增獲得VDAG_7169基因敲除載體片段B(即2/3G418+VDAG_07169-3F片段);
2、PEG介導的原生質體轉化
使用PEG介導的遺傳轉化方法,將構建的VDAG_07169敲除片段VDAG_07169-5F+2/3G418,2/3G418+VDAG_07169-3F導入本發明方法制備的VdXS11-△VdKu80-9敲除突變體菌株的原生質體內,獲得VDAG_07169基因敲除的待選轉化子,具體轉化方法參考(Goswami,R S.Targeted gene replacement in fungi using a split-marker approach.Methods Mol Biol,2011,835:255-269)的方法進行。
3、VDAG_07169敲除突變體篩選和統計
將步驟2中獲得的VDAG_07169基因敲除的待選轉化子轉接至含有50μg/ml Geneticin的PDA抗性平板培養基中,于室溫條件下黑暗培養7天,用1ml蒸餾水沖洗菌落,收集分生孢子;接著將少量分生孢子懸浮液均勻的涂布到含有50μg/ml Geneticin的PDA抗性平板培養基上進行單胞純化,挑取單胞轉化子;然后將純化后的轉化子分生孢子進行甘油保存并采用CTAB法提取基因組DNA;再接著以提取的轉化子DNA為模板,以VDAG_07169-IF/VDAG_07169-IR為引物篩選VDAG_07169基因敲除突變體,并統計敲除突變體的比例(敲除突變體占所有篩選轉化子的比例),本試驗例中所使用的引物如表1所示;統計結果如表2所示。
表2野生型菌株和△VdKu80突變體菌株基因敲除率的對比
為了驗證VdXS11-△VdKu80-9敲除突變體菌株確實具有顯著提高基因敲除效率的特性,本發明選取了兩個基因VDAG_00736和VDAG_07169作為敲除對象。本發明成功構建了VDAG_00736和VDAG_07169融合Geneticin的敲除片段,經過轉化成功獲得了與用野生型大麗輪枝菌菌株轉化相當的轉化子數量。
分別使用VDAG_00736和VDAG_07169基因特異性引物,篩選使用野生型菌株和使用VdXS11-△VdKu80-9敲除突變體菌株獲得的具有遺傳霉素抗性的轉化子。結果表明使用野生型菌株進行轉化獲得的轉化子,均沒有篩選得到VDAG_00736和VDAG_07169敲除突變體,敲除效率接近為0。而使用VdXS11-△VdKu80-9敲除突變體菌株進行轉化獲得轉化子,其基因敲除率大約為20%-25%,其中VDAG_00736的敲除效率為25%,VDAG_07169的敲除效率為21.4%(表2)。
研究表明,本發明獲得的大麗輪枝菌VdKu80敲除突變體菌株能夠顯著提高基因敲除率,同時敲除VdKu80對于大麗輪枝菌生長、產孢、微菌核形成、致病性等表型無顯著影響。因此本發明獲得的大麗輪枝菌VdKu80敲除突變體菌株對于后續的基因功能研究具有巨大的實用價值。
- 還沒有人留言評論。精彩留言會獲得點贊!