基于信號譜差異的混合樣本單核苷酸多態性的檢測方法與流程
本發明涉及一種單核苷酸多態性的檢測方法,尤其涉及一種基于信號譜差異的混合樣本單核苷酸多態性的檢測方法。
背景技術:
單核苷酸多態性(Single nucleotide polymorphism,SNP)是群體基因組中單個核苷酸位置上發生的突變。目前,SNP是使用最廣泛的分子標記之一。以人類基因組為例,單核苷酸多態性占遺傳突變的絕大部分,而且可能是決定個體差異的主要原因。據統計,人類基因組中大約每1000-2000個核苷酸中就有一個SNP,SNP的高豐度、易于檢測等特點決定了它非常適用于對復雜性狀與疾病的遺傳研究以及基于群體的基因識別的研究等方面。
SNP已經成為鑒定遺傳因子的一種重要工具并在許多領域得到了實際應用,例如法醫鑒定、臨川診斷、動植物育種以及改善生物分子產量等。隨著高通量測序技術的發展,對SNP進行大規模鑒定分析在許多物種中已經變得可行。然而,從高通量測序數據中檢測SNP通常會受到測序錯誤以及測序片段比對錯誤的影響。因此,在實際應用之前,應該對發現的SNP進行進一步的驗證以篩選出真正的SNP。
現有技術中有一種基于單核苷酸添加焦磷酸測序技術,從混合樣本中檢測SNP并計算其比例的方法。該方法基于正態性檢驗和動態規劃算法,輸入純野生型樣本和混合樣本的焦磷酸測序信號譜,能夠檢測出SNP的位置、比例以及突變核苷酸。該方法的一個缺點是,在焦磷酸測序過程中,核苷酸的添加過程需要經過嚴格設計以保證野生型序列與突變型序列的延伸節奏不一致。
技術實現要素:
發明目的:針對以上問題,本發明提出一種基于信號譜差異的混合樣本單核苷酸多態性的檢測方法。
技術方案:為實現本發明的目的,本發明所采用的技術方案是:一種基于信號譜差異的混合樣本單核苷酸多態性的檢測方法,包括以下步驟:
第一步:提取野生型樣本的DNA和混合樣本的DNA,進行聚合酶鏈式反應,得到單鏈PCR產物,其中,混合樣本包括野生型序列和突變型序列;
第二步:對野生型樣本和混合樣本的單鏈PCR產物進行三輪循環的實時合成測序,獲得三組信號譜;
第三步:根據三輪測序實驗獲得的野生型樣本與混合樣本的信號譜,計算野生型樣本與混合樣本的信號譜之間的差異,利用枚舉方法推斷混合樣本中可能存在的單核苷酸多態性位點;
第四步:綜合分析三輪測序實驗所檢測出的可能的單核苷酸多態性位點,進行一致性判斷,判斷混合樣本中是否存在單核苷酸多態性位點,如果混合樣本中含有單核苷酸多態性位點,則歸納出單核苷酸多態性位點的位置、突變核苷酸及突變核苷酸的比例。
第三步中利用枚舉方法推斷混合樣本中可能存在的單核苷酸多態性位點的步驟如下:
(1)定義每輪測序實驗獲得野生型樣本的信號譜為W=(W1,W2,…,Wn),其中,Wi為野生型樣本第i次添加兩核苷酸時的信號強度;混合樣本的測序信號譜為P=(P1,P2,…,Pn),其中,Pi為混合樣本第i次添加兩核苷酸時的信號強度;
(2)定義突變型序列的信號譜為M=(M1,M2,…,Mn),其中,突變序列的比例為a,則P=(1-a)W+aM;
(3)實際測序試驗中,Wi、Pi服從正態分布:其中,CV為信號強度的變異系數,和為信號強度的理論值;
(4)定義差別譜D為混合樣本的信號譜P與野生型樣本的信號譜W之差,即D=P-W,則結合步驟(2)可得D=a(M-W),枚舉野生型序列與突變型序列信號譜之間的差別,計算混合樣本中突變序列的比例a;
(5)結合步驟(3)和(4)可得定義Sp來評估所枚舉的野生型序列與突變型序列信號譜之間每種差別的可能性,且選擇最大Sp值時的差別譜作為野生型序列與突變型序列信號譜之間的差別譜,其中:
(6)野生型序列與突變型序列信號譜之間存在五種差別類型:Type-0、Type-1、Type-2、Type-3和Type-4,假設野生型序列與突變型序列之間的不一致延伸發生在第j步循環測序反應過程,定義S為第j步循環測序反應中一致延伸的核苷酸個數,定義函數f(Wj)為根據信號強度Wj計算的延伸核苷酸的個數,可以得出S的表達式和單核苷酸多態性位點的位置L的表達式分別為:
其中,為突變序列的比例的平均值;
(7)結合步驟(6)檢測出的單核苷酸多態性位點的位置L和加入的兩核苷酸信息得到突變核苷酸的信息。
第四步中綜合分析進行一致性判斷的步驟如下:
(1)定義Sc來評價野生型樣本的信號譜與混合樣本的信號譜之間的一致性,同時定義Sm=Sc-Sp;當突變序列存在時,Sc的值大于Sp,突變序列的比例越高,Sm的值越大;其中:
(2)根據三輪測序實驗結果,定義具有最低Sm值的一輪測序實驗中的野生型序列與突變型序列的信號譜完全一致,即野生型序列與突變型序列信號譜之間的差別類型為Type-0;
(3)根據另兩輪測序實驗確定的核苷酸突變位置和突變核苷酸信息是否一致來確定單核苷酸多態性;如果兩次推斷出的單核苷酸多態性位點的位置一致,則判定混合樣本中包含有突變序列,并以兩次計算的突變序列比例的平均值作為突變比例的比;根據兩次推斷出的可能的突變核苷酸的集合,取其交集作為檢測出的突變核苷酸;如果兩次推斷出的單核苷酸多態性位點的位置不一致,則判定混合樣本中不包含單核苷酸多態性。
有益效果:本發明相比于現有技術,具有更高的準確度;兩核苷酸的循環添加順序不需要經過復雜的實驗設計,實驗更容易操作;當混合樣本中不包含單核苷酸多態性時,具有更好的魯棒性以及更低的假陽性發現率;允許野生型序列未知,并能夠確定單核苷酸突變位點的位置、突變核苷酸及其比例;成本低廉。
附圖說明
圖1是本發明所述的檢測方法的邏輯流程圖;
圖2是進行兩核苷酸實時合成測序所獲得的三組信號譜;
圖3是五種信號譜差別類型;
圖4是三輪測序中,野生型樣本與混合樣本的信號譜。
具體實施方式
下面結合附圖和實施例對本發明的技術方案作進一步的說明。
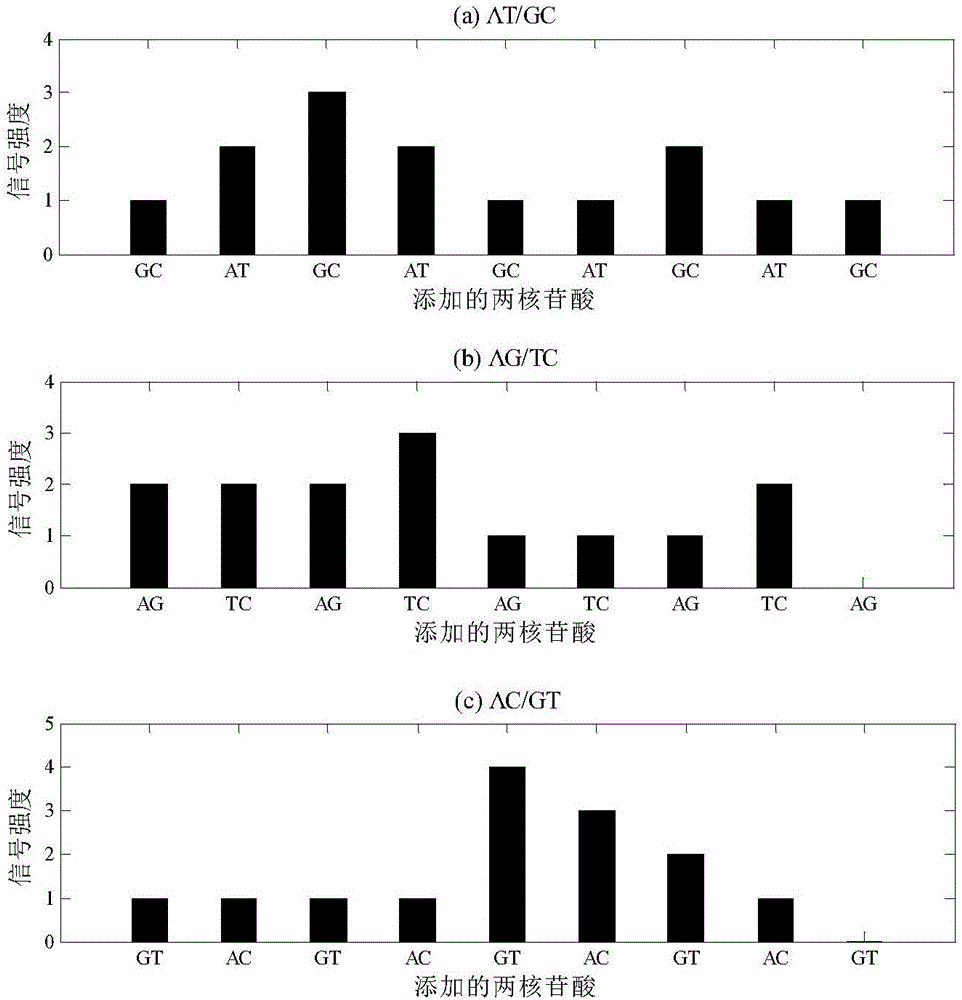
如圖1所示是本發明所述的基于信號譜差異的混合樣本單核苷酸多態性的檢測方法,首先提取野生型樣本的DNA和混合樣本的DNA,進行聚合酶鏈式反應,即PCR擴展,得到單鏈PCR產物,利用一種兩核苷酸實時合成技術對野生型樣本和混合樣本分別進行獨立測序獲得各自的信號譜。以待測序列“GATCGGTTCACGTC”為例,對野生型樣本和混合樣本的單鏈PCR產物進行三輪循環的兩核苷酸實時合成測序(A+G)/(C+G)、(A+C)/(G+T)、(A+T)/(C+G),獲得三組信號譜,如圖2所示。
在實際實驗中,信號強度與每次延伸的核苷酸個數成正比,并且可以經過標準化轉換為延伸的核苷酸的個數。為了展示方便,本發明中直接使用延伸的核苷酸的個數來表示信號強度。
記R(reference)為野生型序列在單核苷酸多態性位點處的核苷酸,V(variant)為突變序列在單核苷酸多態性位點處的核苷酸,B(before)為野生型序列在單核苷酸多態性位點前一位的核苷酸,A(after)為野生型序列在單核苷酸多態性位點后一位的核苷酸。理論上,野生型與單堿基突變型序列的測試信號譜之間的差別僅有五種模式,如圖3所示。
在給定一種兩核苷酸添加方案的前提下,以AT/CG為例,定義A與T為一對雙胞胎核苷酸(twin-nucleotide),C與G一對雙胞胎核苷酸,也就是說,突變為A與T之間的突變或者C與G之間的突變,相鄰的雙胞胎核苷酸將會在同一步測序中一起延伸。如果R和V為一對雙胞核苷酸,野生型序列與突變型序列的延伸步驟將完全一致,即信號譜將完全一致,定義這種情況下野生型序列與突變型序列的信號譜之間的差別模式為Type-0。
如果R和V不是雙胞胎核苷酸,同時B和A也不是雙胞胎核苷酸,那么在測序過程中,野生型序列與突變型序列的延伸步驟幾乎完全一致。然而在B與A分別延伸的兩步循環測序過程中,野生型序列與突變型序列所對應的信號強卻存在著差別。具體來說,如果R和B是雙胞胎核苷酸時,此時V和A也將成為雙胞胎核苷酸,在這種情況下,在B延伸的那一步測序過程中,野生型序列所延伸的核苷酸將比突變型序列所延伸的核苷酸多一個,而在A延伸那一步測序過程中,野生型序列所延伸的核苷酸將比突變型序列所延伸的核苷酸少一個。對應的,定義這種情況下野生型與突變型序列的信號譜之間的差別模式為Type-1。反過來,如果R和A是雙胞胎核苷酸時,此時V和B也將成為雙胞胎核苷酸,定義這種情況下野生型與突變型序列的信號譜之間的差別模式為Type-2。
如果R和V不是雙胞胎核苷酸,而B和A是雙胞胎核苷酸,那么在測序過程中,在單核苷酸多態性位點之后,與野生型序列的延伸過程相比,突變型序列的延伸將會領先兩步或者滯后兩步。具體來說,當B和V是雙胞胎核苷酸時,此時B和R將不是雙胞胎核苷酸,因此B、V和A能夠在同一步測序中延伸,而B、R和A卻需要在三步循環測序過程才能延伸完成,所以突變型序列的延伸將會領先兩步。對應的,定義這種情況下野生型序列與突變型序列的信號譜之間的差別模式為Type-3。反過來,當B和R是雙胞胎核苷酸時,此時B和V將不是雙胞胎核苷酸,突變型序列的延伸將會滯后兩步,定義這種情況下野生型與突變型序列的信號譜之間的差別模式為Type-4。在Type-3型和Type-4型這兩種情況下,野生型與突變型序列的信號譜可能在突變位置之后的多步循環測序中存在著差別。
根據三輪測序實驗獲得的野生型樣本與混合樣本的信號譜,針對每種兩核苷酸添加方式,分別計算野生型樣本與混合樣本的信號譜之間的差異,使用枚舉方法推斷混合樣本中可能的單核苷酸多態性位點的位置、比例和突變核苷酸。假設兩核苷酸測序結果中,野生型樣本的信號譜為W=(W1,W2,…,Wn),其中Wi為第i次添加兩核苷酸時的信號強度。類似的,定義突變型序列的信號譜為M=(M1,M2,…,Mn),混合樣本的測序信號譜為P=(P1,P2,…,Pn)。在實際實驗過程中,需要使用延伸核苷酸個數為1的那一步循環測序所對應的峰值對信號譜進行標準化,以消除野生型樣本與混合樣本DNA濃度不一致所帶來的影響。我們假設W和P是標準化之后的信號譜。混合樣本由野生型序列和突變序列組成,其中突變序列的比例為a。理論上滿足:
P=(1-a)W+aM 式1
研究顯示,Wi的值實際上服從正態分布,如式2所示,類似的,混合樣本的信號譜中,第i次添加兩核苷酸時的信號強度Pi同樣服從正態分布,如式3所示。
其中,和為信號強度的理論值,即理論上延伸的核苷酸個數。CV為信號強度的變異系數,能夠直接反應焦磷酸測序實驗的精準度。CV的具體值為標準差除以均值。在實際中,CV的值取決于測序實驗的準確度。
在給定野生型樣本的信號譜W和混合樣本的信號譜P的前提下,定義差別譜D為:
D=P-W 式4
根據式1和式4可以推斷出:
D=a(M-W) 式5
同樣地,在第i次循環測序中,Di的值同樣服從一個正態分布,基于式2-式5,可以推斷出,Di服從如下分布:
在給定野生型樣本信號譜W與混合樣本信號譜P的前提下,使用枚舉方法來推測突變發生的位置。具體來說,根據野生型序列與突變型序列信號譜之間的五種差別類型,假設野生型序列與突變型序列中不一致的延伸可能開始于任意一步循環測序中,枚舉出野生型序列與突變型序列信號譜之間所有可能的差別譜。因為D是觀察值并且是已知的,所以根據式5能夠計算出混合樣本中突變序列的比例。由于野生型序列與突變型序列信號譜在至少兩步測序過程中存在著差別,所以會獲得多個突變比例的計算值,取其平均值為最終的突變序列比例的計算值,并記為
定義一個得分Sp來評估所枚舉的野生型序列與突變型序列信號譜之間每種差別的可能性,如式7所示。
其中,n為循環測序的步數。簡而言之,Sp為差別譜中的每個元素對應的標準分(也被稱為Z-score)的平均值。所以,Sp能夠反應在假設的野生型序列與突變型序列信號譜的差別的前提下,觀察值D與理論值P-W之間的偏差程度。對應地,較低的Sp值表示觀察值D與理論值P-W之間的偏差較小,即所假設的野生型序列與突變型序列信號譜之間的差別譜具有更高的概率。
由于野生型序列和真實的突變比例a是未知的,導致和也都是未知的,根據式4和式5,可以推斷出P-W等于a(M-W),在給定假設的野生型序列與突變型序列信號譜之間的差別譜,即M-W的前提下,可以使用來近似類似的,使用來近似此外,CV是一個取決于測序實驗準確度的常量,所以修改Sp為式8,該值仍然能夠反映觀察值D與理論值P-W之間的偏差程度。
在給定野生型樣本信號譜W和混合樣本信號譜P的前提下,基于野生型序列與突變型序列信號譜之間的五種差別類型,枚舉出野生型序列與突變型序列信號譜之間所有可能的差別譜,并計算突變型序列的比例。同時,對每種假設的差別譜進行打分,獲得Sp值,該值能夠間接反映各假設的差別譜的可能性。最終,選擇具有最大Sp值的差別譜作為野生型序列與突變型序列信號譜之間可能的差別譜。
給定野生型序列與突變型序列信號譜之間差別譜,可以推斷出單核苷酸多態性位點的位置為測序過程中野生型序列與突變型序列之間不一致延伸所開始的地方。假設不一致延伸發生在第j步循環測序反應過程中。顯而易見,在第j-1步循環測序反應中所一致延伸的核苷酸個數能夠直接根據野生序列的信號譜推斷出來。而在第j步循環測序反應中所一致延伸的核苷酸個數,記為S,為和之間的較小值。理論上,如果差別譜屬于Type-1型或Type-4型,S等于而如果差別譜屬于Type-2型或Type-3型,S等于
對于Type-1型差別譜,的值等于而對于Type-4型差別譜,的值則可以通過式9計算得到。
因此,可以得出S的表達式,如式10,而最終計算的單核苷酸多態性位點的位置L的表達式,如式11。
由于野生型序列式未知的,所以在每步循環測試反應中真實延伸的核苷酸個數也是未知的。在實際的實驗中,真實延伸的核苷酸個數是與信號強度成正相關但并非嚴格相等,而且每個對應的信號強度是處于一定的取值范圍內的。反過來,給定信號強度,可以根據其值所處的區間范圍直接推斷出真實延伸的核苷酸個數。在這里,使用函數f(Wi)來標記根據信號強度Wi所推斷而獲得的核苷酸個數。
因此,式10可以修改為:
在式13中,使用來近似突變比例a的值,并使用Pj-Wj來近似的值。綜合,最終推測出單核苷酸多態性位點的位置L如式14。
同時,推斷出突變型序列與野生型序列在單核苷酸多態性位點處的可能核苷酸。假設不一致延伸發生在第j步循環測序反應過程中。對于Type-2和Type-3型差別譜,野生型序列在單核苷酸多態性位點處的可能核苷酸即為在第j+1步循環測序過程中添加的兩種核苷酸,而突變序列在單核苷酸多態性位點處的可能核苷酸即為在第j步循環測序過程中添加的兩種核苷酸。與此相反的是,對于Type-1和Type-4型差別譜,野生型序列在單核苷酸多態性位點處的可能核苷酸即為在第j步循環測序過程中添加的兩種核苷酸,而突變序列在單核苷酸多態性位點處的可能核苷酸即為在第j+1步循環測序過程中添加的兩種核苷酸。
綜合分析三輪測序實驗所檢測出的可能的單核苷酸多態性位點,進行一致性判斷,如果混合樣本中含有單核苷酸多態性位點,歸納出其位置、突變核苷酸及其比例。
兩核苷酸實時測序技術中,野生型樣本和混合樣本的信號譜均有三組,分別對應三種不同的兩核苷酸組合添加方案。理論上,在三輪測序實驗中,必定有一輪測序中,參考核苷酸R與突變核苷酸V屬于一對雙胞胎核苷酸,即野生型樣本和混合樣本的信號譜是完全一致的。因此給定三輪測序實驗結果,首先要鑒定出在哪一輪測序實驗中,野生型樣本的信號譜與混合樣本的信號譜是完全一致的。
當核苷酸R與核苷酸V屬于一對雙胞胎核苷酸時,野生型樣本的信號譜與混合樣本的信號譜是完全一致的,即差別譜中的每個元素的值均為0,與式8類似,定義一個打分Sc,如式15,來評價野生型樣本的信號譜與混合樣本的信號譜之間的一致性。
其中,n為循環測序的步數。具體而言,Sc是Sp的一個特殊情況,即假設的野生型序列信號譜與突變型序列信號譜的差別譜中所有的元素的值均為0。
當假設突變不會導致野生型序列和突變型序列具有不同的信號譜時,Sc可以反應觀測到的差別譜D與理論上的差別譜P-W的偏差程度,而當假設突變會導致野生型序列和突變型序列具有不同的信號譜時,Sp可以反應觀測到的差別譜D與理論上的差別譜P-W的偏差程度。最終,定義Sm,如式16,來評估混合樣本中存在著突變型序列的可能性。
Sm=Sc-Sp 式16
當突變序列存在時,Sc的值會比Sp更大。而且突變序列的比例越高,Sm的值將會更大。直觀上,Sm能夠反映突變序列的存在對野生型樣本信號譜與混合樣本信號譜之間的一致性的影響。因此推測具有最低Sm值的一輪測序實驗中,野生型序列與突變型序列的信號譜完全一致。
在三輪測序完成之后,針對每輪測序實驗的結果,分別利用枚舉方法檢測可能的單核苷酸多態性位點,同時計算對應的Sm。認定具有最低Sm的那一輪測序實驗中,野生型樣本和混合樣本的信號譜是完全一致的。那么,在該輪測序實驗中,參考核苷酸R與突變核苷酸V為一對雙胞胎核苷酸。
根據另外兩輪測序實驗中所檢測出來可能的單核苷酸多態性位點的位置,進行一致性判斷。如果兩次推斷出的單核苷酸多態性位點的位置一致,則認為混合樣本中包含有突變序列,并以兩次計算的突變序列比例的平均值作為突變比例的最終計算值,根據兩次推斷出的可能的突變核苷酸的集合,取其交集作為最終檢測出的突變核苷酸。反之,如果兩次推斷出的單核苷酸多態性位點的位置不一致,則認為混合樣本中不包含單核苷酸多態性。
下面以一個實際的測試實驗為例進行說明。
基于信號譜差異推斷擬定的混合樣本中的單核苷酸多態性位點,該混合樣本含野生型序列“CGACCAGCT”和突變型序列“CGATCAGCT”。
(1)將100%野生型序列和由90%的野生型序列與10%的突變型序列組成的混合樣本,獨立經過三輪兩核苷酸實時合成測序,分別模擬獲得野生型樣本與混合樣本的測序信號譜,結果如表1及圖4所示。設定測序過程中,信號變異系數CV值為0.001。
(2)每輪兩核苷酸實時合成測序中每步循環測序反應信號強度的結果如表1所示。
表1
(3)根據式4、5、8、13、14、15、16。針對每輪兩核苷酸測序結果,首先計算野生型樣本與混合樣本的信號譜之間的差異,在此基礎之上,采用枚舉方法推斷可能的單核苷酸多態性位點的位置、突變序列的比例和突變核苷酸,同時計算Sm值,結果如表2所示。
表2
(4)根據步驟(3)中表2的結果,第二輪AG/CT兩核苷酸測序的Sm值在三輪測序中最低,可以確定這一輪測序實驗中,野生型序列與突變型序列信號譜之間的差別類型為Type-0。在第二輪測序中,添加的兩核苷酸分別為AG與CT,據此可以推斷突變為A與G之間的突變或者C與T之間的突變。值得注意的是,根據第二輪測序結果,本發明方法推斷混合樣本中可能存在一個單核苷酸多態性位點,其位于第3位核苷酸處,對應的突變序列比例為0.0043,顯而易見,這是由于測序過程中引入的隨機誤差所導致的。
根據第一輪和第三輪兩核苷酸測序結果獨立推斷出的可能的單核苷酸多態性位點的位置均為第4位核苷酸,因此可以判定混合樣本中包含有突變序列、并確定單核苷酸多態性位點發生在第4位核苷酸處。根據第一輪和第三輪測序計算混合樣本中的突變序列的比例分別為0.1006和0.0999,因此最終計算出混合樣本中突變序列的比例為0.10025。
根據第一輪測序結果,確定野生型序列在單核苷酸多態性位點處核苷酸可能為C或G,而根據第三輪測序結果,本發明方法檢測出野生型序列在單核苷酸多態性位點處核苷酸可能為A或C,因此可以歸納出野生型序列在單核苷酸多態性位點處核苷酸為C。
根據第一輪測序結果,確定突變序列在單核苷酸多態性位點處核苷酸可能為A或T,而根據第三輪測序結果,本發明方法檢測出突變序列在單核苷酸多態性位點處核苷酸可能為G或T,因此可以歸納出突變序列在單核苷酸多態性位點處核苷酸為T。
- 還沒有人留言評論。精彩留言會獲得點贊!
- CYP2C9*3基因多態性快速檢測的引物、分子信標、試劑盒及其檢測方法與流程
- 一種單核苷酸多態性位點在輔助鑒定大豆花葉病毒抗性中的應用的制造方法與工藝
- DHFR基因多態性檢測體系及其試劑盒的制造方法與工藝
- ERCC1基因多態性檢測體系及其試劑盒的制造方法與工藝
- 一種判定中國人群rs125055單核苷酸多態性與兒童IBD之間的相關性的方法與制造工藝
- 黃牛ADNCR基因單核苷酸多態性的四引物擴增受阻突變體系PCR檢測方法及其應用與制造工藝
- 使用PNA探針的熔解曲線分析、使用熔解曲線分析用于分析核苷酸多態性的方法和試劑盒與制造工藝
- 人KRAS中單核苷酸多態性的檢測的制造方法與工藝
- 一種InDel位點基因型分型的方法與制造工藝
- 單核苷酸多態性rs145562243在篩查麻風患者中的應用的制造方法與工藝
- 一種用于評估皮膚防御情況的引物和探針及試劑盒的制作方法
- 一種用于評估皮膚防曬情況的引物和探針及試劑盒的制作方法
- 一種用于評估皮膚抗皺情況的引物和探針及試劑盒的制作方法
- 人PLIN1基因rs2289487位點多態性檢測方法及試劑盒的制作方法
- 測定人NEFM基因rs12515位點多態性的方法及試劑盒的制作方法
- 一種人TERF1基因rs3863242位點多態性檢測技術的制作方法
- 測定人CCNH基因rs3093816位點多態性的方法及試劑盒的制作方法
- 測定人NEFL基因rs2979704位點多態性的方法及試劑盒的制作方法
- 測定人PON1基因rs854560位點多態性的方法及試劑盒的制作方法
- 測定人PON1基因rs662位點多態性的方法及試劑盒的制作方法
- CYP2C9*3基因多態性快速檢測的引物、分子信標、試劑盒及其檢測方法與流程
- 一種單核苷酸多態性位點在輔助鑒定大豆花葉病毒抗性中的應用的制造方法與工藝
- DHFR基因多態性檢測體系及其試劑盒的制造方法與工藝
- ERCC1基因多態性檢測體系及其試劑盒的制造方法與工藝
- 一種判定中國人群rs125055單核苷酸多態性與兒童IBD之間的相關性的方法與制造工藝
- 黃牛ADNCR基因單核苷酸多態性的四引物擴增受阻突變體系PCR檢測方法及其應用與制造工藝
- 使用PNA探針的熔解曲線分析、使用熔解曲線分析用于分析核苷酸多態性的方法和試劑盒與制造工藝
- 人KRAS中單核苷酸多態性的檢測的制造方法與工藝
- 一種InDel位點基因型分型的方法與制造工藝
- 單核苷酸多態性rs145562243在篩查麻風患者中的應用的制造方法與工藝
- 用于檢測人類CYP2D6基因多態性的核酸、試劑盒及方法與流程
- 單核苷酸多態性位點rs2523607在篩查皰疹樣皮炎患者中的應用的制作方法與工藝
- 核苷酸多態性檢測方法與流程
- 單核苷酸多態性rs146466242在篩查麻風患者中的應用的制作方法與工藝
- 基于信號譜差異的混合樣本單核苷酸多態性的檢測方法與流程
- 一種檢測牛PCAF基因單核苷酸多態性的RFLP方法及試劑盒與流程
- 秦川牛microRNA?320a?1基因單核苷酸多態性的檢測方法及其應用與流程
- 單核苷酸多態性rs76418789在篩查麻風患者中的應用的制作方法與工藝
- 判定rs4676410和rs2531875單核苷酸多態性與AS之間相關性的方法與流程
- 一種可視化檢測基因序列中單核苷酸多態性位點的反向探針、試劑盒及其檢測方法與流程