一種PDE4抑制劑氯比普蘭的制備方法與流程
本發明涉及一種PDE4抑制劑的制備方法,具體為化合物氯比普蘭的制備方法。
背景技術:
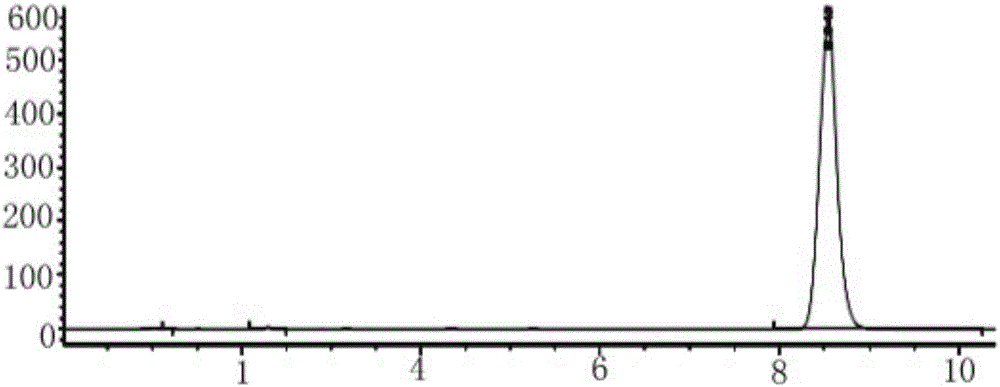
:磷酸二酯酶4(PDE4)與中樞神經系統和免疫系統的功能有密切的關系,其抑制劑被認為是作用于細胞內靶點的新抗炎藥和中樞神經系統候選藥物。第一代PDE4抑制劑如咯利普蘭具有很好的抗抑郁效果,導致其不能上市的原因是抑制PDE4所帶來的惡心嘔吐等副反應。與第一代PDE4抑制劑相比,第二代PDE4抑制劑具有更好的治療效果,但至今還沒有能避免嘔吐的PDE4抑制劑上市。開發出能夠避免惡心嘔吐反應的新型PDE4抑制劑在抑郁癥和認知障礙的治療上具有很好的應用前景。CN201210037980.3號中國專利公開了一系列的PDE4抑制劑,并證明氯比普蘭(chlorbipram)在對東莨菪堿誘導的學習記憶獲得性障礙大鼠模型具有很好的改善作用;對強迫游泳和懸尾行為絕望模型小鼠也具有良好的抗抑郁活性,并且已采用比格犬證明其僅有微弱甚至無致嘔吐惡心副作用。該專利也同時公開了氯比普蘭(chlorbipram)的合成方法,該方法采用甲基溴化后與NH類化合物對接合成制得,反應路線見Scheme1。由于NBS溴化的副反應多,溴化物不穩定,對接反應收率較低,其合成有一定的局限性,不適合該化合物的后續研發。Scheme1技術實現要素:本發明公開了一種PDE4抑制劑氯比普蘭的制備方法,實驗證明,該方法的產率高達71%。本制備方法的合成路線為:包括以下步驟:(1)以3-溴-4-甲氧基苯甲醛為起始原料A,還原反應生成化合物B,即3-溴-4-甲氧基苯甲醇;(2)將化合物B與間氯苯硼酸,在鈀鹽催化劑的催化下,進行Suzuki反應,生成化合物C;(3)將化合物C用氯化試劑進行氯化,生成化合物D;(4)將原料化合物A1與3,6-二氯噠嗪,加熱至回流反應,生成化合物A2;化合物A2與醋酸鈉加熱回流反應,生成化合物A3;(5)將化合物D和化合物A3在堿性條件下回流反應,生成目標產物E氯比普蘭。步驟(1)所述的還原反應采用的還原劑優選NaBH4。步驟(2)所述的鈀鹽催化劑優選四三苯基膦鈀或醋酸鈀或醋酸鈀和配體尿素。四三苯基膦鈀與化合物B的投料比優選1:20,催化劑醋酸鈀與化合物B的投料比優選1:30。步驟(3)所述的氯化試劑優選四氯化碳/三苯基膦或三聚氯氰/二甲基甲酰胺體系。步驟(5)所述的回流反應溶劑優選乙二醇二甲醚或二甲基甲酰胺。本發明的制備方法相對于現有技術,具有以下優點:產率高達64-71%,較之前的合成方法產率提高42.2%-57.8%,且原料易得價廉,反應條件溫和,對設備要求低,可實現產業化生產。本發明為氯比普蘭的合成方法提供了一條高效易行的新途徑。說明書附圖圖1是本實施例制備的目標化合物氯比普蘭的HPLC結果圖。具體實施方式下面結合實施例對本發明的制備方法作進一步的說明。但本發明的保護的范圍不以以下實施例為限。實施例1(1)取30g原料A,3-溴-4-甲氧基苯甲醛,分批次緩慢加入NaBH48-10g,在乙醇中反應。通過TLC監控反應。反應完全后,除去乙醇,萃取,合并有機相,無水硫酸鈉干燥,旋干得白色固體3-溴-4-甲氧基苯甲醇30.0g,產品不需要純化即可進行下步反應,收率>98%。(2)取10g3-溴-4-甲氧基苯甲醇,加入間氯苯硼酸8.6g,碳酸鉀18.8g,四三苯基膦鈀1.0g,異丙醇/水溶劑100mL,室溫攪拌。待固體全部溶解,除去反應體系中的氧氣,無氧反應,加熱,反應完后,減壓旋掉異丙醇和部分水,然后用乙酸乙酯萃取,合并有機層,用無水硫酸鈉干燥后用抽濾擋掉鈀鹽,旋干,得微黃黏稠液體化合物C11.46g,產品不需要純化即可進行下步反應,收率>95%。(3)取化合物C11.5g(按純品折算),加四氯化碳7.4g,分批加入三苯基膦11.0g,在二氯甲烷中,常溫攪拌,TLC檢測反應。反應完后,濃縮反應液,柱分離除雜,得無色透明油狀液體化合物D,11.1g,收率90%。(4)加31.2g化合物A1與28.2g3.6-二氯噠嗪在乙醇中,加熱至回流,TLC監控反應,反應結束后,減壓旋掉乙醇得粗品,重結晶,得黃色固體化合物A244g,收率84.3%。取50g上述得到的化合物A2用醋酸/醋酸鈉,攪拌加熱回流,TLC檢測。反應結束后除去醋酸鈉,減壓除去乙酸,用乙醇重結晶的淡黃色固體化合物A3,收率92%。(5)取11.0gA3于100mL圓底燒瓶中,加入11.0g化合物D,14.5g無水碳酸鉀,溶劑乙二醇二甲醚,回流。反應完后,用乙酸乙酯/石油醚為洗脫劑,柱分離,得白色固體化合物E,即氯比普蘭15g,收率75%。經上述方法,計算得總收率為67.5%。實施實例2:在實施例1的基礎上,其他步驟不變,實施例1中步驟(2)方法變為以下方案:取10g化合物B,3-溴-4-甲氧基苯甲醇,加入間氯苯硼酸8.6g,碳酸鉀18.8g,尿素82.8mg,醋酸鈀210mg,異丙醇/水溶劑100mL,室溫攪拌。待固體全部溶解,除去反應體系中的氧氣,無氧反應,加熱,反應完后,減壓旋掉異丙醇和部分水,然后用乙酸乙酯萃取,合并有機層,用無水硫酸鈉干燥后用抽濾擋掉鈀鹽,旋干,得微黃黏稠液體化合物C,11.46g,收率>95%。本實施例的總收率為65.5%。實施實例3:在實施例1的基礎上,其他步驟不變,實施例1中步驟(3)方法變為以下方案:8.13g三聚氯氰,緩慢攪拌加入DMF,室溫攪拌半小時左右,加入干燥的二氯甲烷,攪拌均勻,加入原料C11.5g,室溫攪拌4-8小時。反應完后有機層用飽和NaCl洗滌,干燥有機層,減壓旋干。粗品用柱分離的方法精置,得無色的油狀氯化產物,即化合物D,11.6g(收率95%)。經計算總收率為71%。實施例4:在實施例1的基礎上,其他步驟不變,實施例1中步驟(5)方法變為以下方案:取11.0gA3于100mL圓底燒瓶中,加入11.0g化合物D,14.5g無水碳酸鉀,溶劑二甲基甲酰胺(DMF),回流。反應完后,用乙酸乙酯/石油醚為洗脫劑,柱分離,得白色固體化合物E14.4g,收率72%。經計算總收率為64.8%。經實驗證明:步驟(2)所述的鈀鹽催化劑與化合物B的投料比為1:10-100的范圍內,都可獲得較好的收率,收率在64%以上。由上述實施例方法所得的目標化合物E,數據表征為:ESI-MS:m/z492.6([M+H]);1HNMR(400MHz,CDCl3)δ1.26(s,1H),3.62(t,J=4.8Hz,2H),3.78(s,3H),3.84(s,3H),4.14(t,J=4.8Hz,2H),5.15(s,2H),6.73(d,J=8.0Hz,1H),6.85-6.96(m,6H),7.36-7.38(m,2H),7.40(d,J=8.4Hz,1H),7.49(s,1H).13CNMR(100MHz,CDCl3)δ41.7,54.3,55.7,55.8,67.6,111.3,112.0,111.8,121.0,122.1,126.7,127.0,127.7,128.9,129.2,129.5,130.0,130.8,131.5,133.7,140.0,147.6,147.9,149.7,156.1,and157.9.HPLC結果如圖1及表1所示。表1峰(#)保留時間(min)峰寬(min)峰面積(mAU·s)峰面積(%)11.1610.05115.741170.072322.2830.069216.698510.210238.5420.21597922.4497199.7176總量7944.88939本發明的制備方法與CN201210037980.3號中國專利公開的合成方法的目標產品收率比較見表2。明顯可見,利用本發明方法,以3-溴-4-甲氧基苯甲醛為起始原料經四步反應,最終氯比普蘭的總收率可以達到71%,一般收率也可達64-71%。而CN201210037980.3號中國專利公開的合成方法,收率只有45%。表2本專利保護的合成方法,所顯示的實施實例具有一定的代表性,但是并不僅僅局限在所述的實例方法。尤其是其中Pd催化的偶聯反應,反應條件不局限在Pd(PPh3)4,Pd(OAc)2及加入不同配體,不同堿及溶劑的反應條件;采用芐基醇轉化為芐基氯的方法也不局限在上述實例的CCl4/PPh3和三聚氯氰/DMF兩種反應方式。凡與本發明構思相同的合成方法,都應落入本發明的保護范圍之內。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1