一個逆境誘導啟動子SlWRKY31P的克隆及應用的制作方法
本發明涉及生物技術和植物基因工程領域,具體為一個逆境誘導啟動子slwrky31p的克隆及應用。本發明是一種來源于番茄的高鹽、干旱或水楊酸逆境誘導啟動子slwrky31p的克隆及應用,該啟動子能夠驅動目的基因在高鹽、干旱或水楊酸逆境條件下在植物中高效表達。
背景技術:
啟動子(promoter)是dna鏈上一段能與rna聚合酶結合并能起始mrna合成的序列,主要由核心啟動子區及其上游調控區組成。核心啟動子區包含轉錄起始位點和tata盒結構(rna聚合酶的結合位點處之一,決定轉錄起始的精確性)。上游調控區包括增強轉錄效率的caat盒及應答元件,應答元件結合轉錄因子,調控下游基因表達。
啟動子分為組成型啟動子(constitutivepromoter)和特異性啟動子(specificpromoter)。組成型啟動子能在所有細胞、任何時候進行轉錄;特異性啟動子又可分為組織特異性啟動子和誘導型啟動子。組織特異性啟動子可調控基因只在某些特定的部位或器官中表達,常表現出發育調節特性。誘導特異性啟動子能夠接受逆境條件下的誘導信號,特異啟動應答基因表達,在植物體內產生大量的特異蛋白,從而做出調節反應,抵抗外界環境。
植物逆境是指對植物施加有害影響的環境因子,對植物產生重要影響的逆境主要有缺水、低溫、鹽堿、高溫等非生物逆境,以及病原等生物逆境。無論非生物逆境還是生物逆境都會造成各種農作物產量和質量的下降,因此,培育和推廣抗逆品種是保證作物穩產高產的有效途徑。植物體內多種基因受逆境脅迫誘導,而抗逆基因的表達受啟動子所調控。因此,在抗逆基因工程中,選擇響應逆境脅迫的誘導型啟動子構建植物表達載體,用以驅動脅迫應答基因在植物體內定時、定位、定量表達,是解決植物逆境脅迫問題的一個重要策略,對于研究植物抗逆能力調控具有重要意義。
目前能應用于植物轉基因研究的逆境誘導型啟動子已有報道,但這些報道的多數啟動子都只是受到單一的逆境脅迫誘導,如鹽誘導啟動子,棉花ghnhx1啟動子(楊國棟等,2007)、菊花cddreba啟動子(chenetal.,2012);高溫誘導啟動子,如番茄lemtshsp啟動子(liuetal.,2001)和小麥hvhsp17啟動子(freemanetal.,2011)等。而對于同時受到多種逆境脅迫誘導的誘導型啟動子的克隆和功能研究卻鮮有報道。實際上,在植物的整個生長過程中,不僅僅只受到單一的逆境脅迫,而往往會在不同的生長時期受到不同的逆境的脅迫,甚至在同一生長時期會同時受到多種逆境脅迫的影響,因此單一的逆境脅迫誘導型啟動子在對提高植物綜合抗逆性能(廣譜抗性)的應用中就顯示出一定的局限性,而多種逆境脅迫誘導型啟動子在這方面的應用將更具優勢。
為此,本發明人經過長期研究,發現了一個受多種逆境誘導的啟動子
slwrky31p,具體為一種來源于番茄的受高鹽、干旱或水楊酸逆境誘導的啟動子slwrky31p,在植物基因工程改造和植物綜合抗逆能力的遺傳改良中具有良好的應用前景。
參考文獻
1.楊國棟.棉花耐鹽基因ghnhx1啟動子的克隆及功能分析[d].山東農業大學,2007.
2.cheny,chens,chenf,etal.functionalcharacterizationofachrysanthemumdichrum,stress-relatedpromoter[j].molecularbiotechnology,2012,52(2):161-169.
3.jianl,shonom.molecularcloningthegeneofsmallheatshockproteininthemitochondriaandencoplasmicreticulumoftomato[j].actabotanicasinica,2001,43(2):138-145.
4.freemanj,sparksca,westj,etal.temporalandspatialcontroloftransgeneexpressionusingaheat-induciblepromoterintransgenicwheat.[j].plantbiotechnologyjournal,2011,9(7):788.
技術實現要素:
本發明的發明目的在于:提供一個受多種逆境誘導啟動子slwrky31p的克隆及其應用。本發明的slwrky31p對通過啟動目的基因在高鹽、干旱或水楊酸逆境條件下的高表達,來提高轉基因植物對高鹽、干旱或病原菌的耐受性和抗性,具有重要意義。
為了實現上述目的,本發明采用如下技術方案:
一種逆境誘導啟動子slwrky31p,其核苷酸序列包括如下(a)或(b)或(c)的序列:
(a)具有seqidno:1所示的核苷酸序列;或
(b)與a)限定的核苷酸序列具有75%以上一致性,且具有啟動子功能的dna分子;或
(c)在高嚴謹條件下與(a)或(b)限定的核苷酸序列雜交且具有啟動子功能的dna分子。
本發明還公開了一種包含所述dna分子的表達盒。
并公開了一種包含所述表達盒的重組載體;
所述重組載體為雙元載體、共合載體,所述重組載體優選
為pbi121sx-slwrky31p。
以及,公開了一種包含所述重組載體的重組微生物,所述重組微生物優選為大腸桿菌或根癌農桿菌eha105。
以及,公開了一種包含所述重組載體的轉基因細胞系。
本發明公開了一種引物對,所述引物對用于擴增上述誘導啟動子,所述引物對為:
正向引物:5’-actagtctgggatatatgatgcttattag-3’
反向引物:5’-ctcgagtaaaagaaagaagatacaagagtg-3’。
本發明具體公開了一種提取、擴增番茄誘導啟動子slwrky31p的pcr方法,包括以下步驟:
1)提取番茄ac+的基因組dna;
2)以番茄ac+的基因組dna為模板,使用引物,利用高保真酶primestarhs,擴增slwrky31p啟動子;
其中,擴增總體積50μl,其中正向引物1μl,反向引物1μl,dna模板1μl,primestarhs25μl,ddh2o22μl。擴增程序為:98℃10秒,55℃5秒,72℃5秒,30個循環;
所述引物為:
正向引物:5’-actagtctgggatatatgatgcttattag-3’
反向引物:5’-ctcgagtaaaagaaagaagatacaagagtg-3’。
進一步的,本發明公開了逆境誘導啟動子slwrky31p在植物中表達的應用;
優選的,所述逆境誘導啟動子slwrky31p在高鹽、干旱環境和/或病原菌侵襲條件下誘導啟動目的基因在植物中表達的應用;
所述植物為雙子葉植物。
所述雙子葉植物為番茄、水稻、擬南芥和/或煙草。
為實現上述目的,本發明提供了一個逆境誘導啟動子slwrky31p的克隆及其應用,本發明中的啟動子,名稱為slwrky31p,來源于番茄,是如下a)、b)或c)的dna分子:a)核苷酸序列是seqidno.1的dna分子;b)與a)限定的核苷酸序列具有75%或75%以上一致性,且具有啟動子功能的dna分子;c)在高嚴謹條件下與a)或b)限定的核苷酸序列雜交,且具有啟動子功能的dna分子。其中,seqidno.1由1849個核苷酸組成,具體如下:
所述高嚴謹條件是在2×ssc(檸檬酸鈉),0.1%sds(十二烷基硫酸鈉)的溶液中,在68℃下雜交并洗膜2次,每次5min;然后用0.5×ssc,0.1%sds的溶液中,在68℃下雜交并洗膜2次,每次15min。
本領域普通技術人員可以很容易地采用已知的方法,例如定向進化和點突變的方法,對本發明的啟動子核苷酸序列進行突變。那些經過人工修飾的,具有與本發明分離得到的啟動子核苷酸序列75%或者更高同一性的核苷酸,只要保持了表達靶基因的啟動子活性,均是衍生于本發明的核苷酸序列并且等同于本發明的序列。這里使用的術語“同一性”指與天然核酸序列的序列相似性。“同一性”包括與本發明的啟動子核苷酸序列具有75%或更高,或85%或更高,或90%或更高,或95%或更高同一性的核苷酸序列。同一性可以用肉眼或計算機軟件進行評價。使用計算機軟件,兩個或多個序列之間的同一性可以用百分比(%)表示,其可以用來評價相關序列之間的同一性。
同時,基于本發明的逆境誘導啟動子,含有slwrky31p的表達盒、重組載體、重組微生物或轉基因細胞系也屬于本發明的保護范圍。
在所述表達盒中,啟動子slwrky31p的下游連接結構基因、或調節基因、或結構基因或調節基因的反義基因、或者能夠干擾內源基因表達的小rna的編碼dna,用于驅動結構基因、或調節基因、或結構基因或調節基因的反義基因、或者天然小rna或人工合成的小rna的表達。
所述表達盒可由所述啟動子slwrky31p、所述啟動子slwrky31p啟動表達的目的基因以及轉錄終止序列組成;所述啟動子以功能性方式與所述目的基因連接,且所述目的基因與所述轉錄終止序列連接。在本發明的一個實施例中,目的基因具體為β-葡萄糖苷酶(β-glucuronidase,gus)基因。
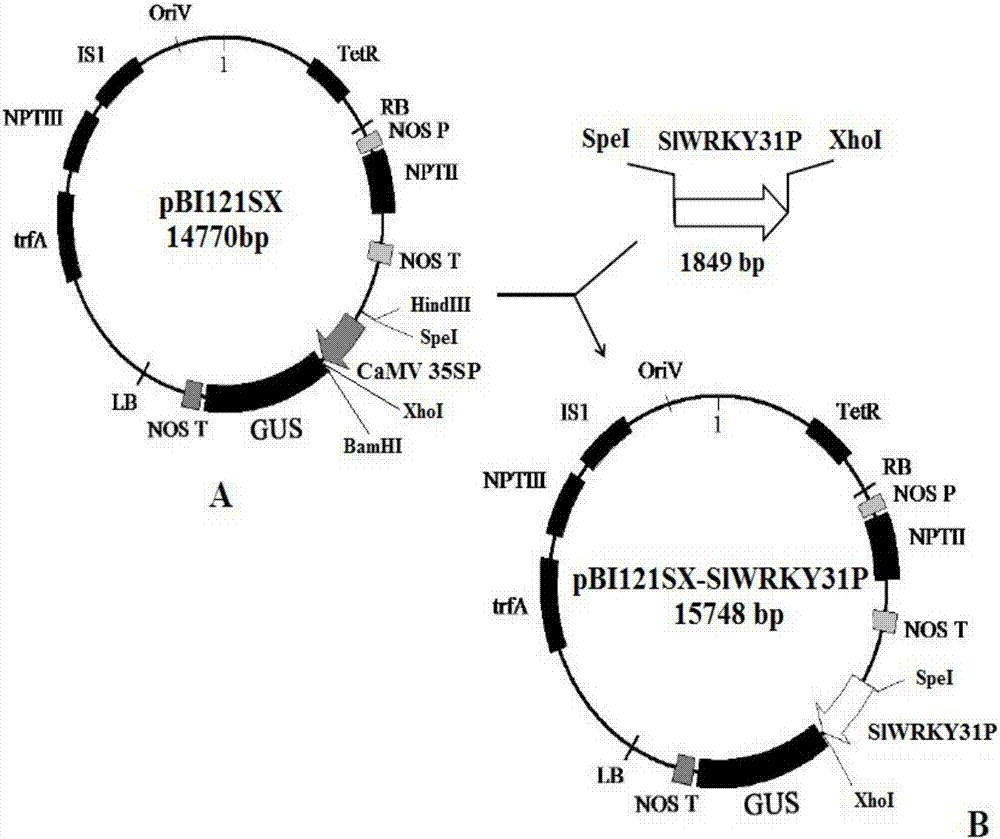
所述重組載體可為含有上述表達盒的重組載體,包括但不限于質粒和病毒。所述重組載體也可為重組植物表達載體。所述重組植物表達載體含有上述表達盒并且能夠將所述的表達盒轉送進入植物宿主細胞、組織或器官及其后代并且能夠或者至少方便所述的表達盒整合宿主的基因組中,它包括但不限于雙元載體、共合載體。所述宿主細胞、組織或器官及其后代是指所有植物細胞或植物組織或植物器官或由這些細胞、組織或器官通過組織分化或無性胚再生并且發育成熟的整體植株(包括種子)。在本發明的一個實施例中,所述重組載體具體為將pbi121sx(植物表達載體pbi121sx是通過在pbi121載體的hindiii位點后面加入了spei位點,在bamhi位點前面加入了xhoi改造而成)的35s啟動子替換為所述啟動子得到的重組載體。所述目的基因具體為β-葡萄糖苷酶基因(β-glucuronidase,gus)。
本發明在slwrky31p啟動子后連接gus報道基因的轉基因實驗,驗證了slwrky31p啟動子受到干旱、高鹽和病原菌侵襲模擬條件的誘導,從而啟動gus基因的表達。通過測定gus基因的表達產物(β-葡萄糖苷酶)的活性,判斷本發明的slwrky31p啟動子的確受到干旱、高鹽或水鹽酸的誘導。
在slwrky31p啟動子后連接的目的基因,理論上來講,可以是任何一個基因,其中包括相關的抗逆基因。slwrky31p啟動子受到干旱、高鹽或水鹽酸的誘導,其后連接的目的基因會受到相應的誘導表達,因為基因的時空表達模式的主要是由啟動子決定。
實驗中的gus基因是驗證本發明slwrky31p啟動子功能的一個常用報道基因,在實際應用中slwrky31p啟動子后面連接的目的基因可以是任意的一個基因,這里目的基因不唯一,可以任意組合。
所述重組表達載體可以通過使用ti質粒、ri質粒、植物病毒載體、直接dna轉化、顯微注射、電導、農桿菌介導或基因槍等常規生物學方法轉化植物器官或組織或細胞,得到轉基因植物細胞或組織或器官及由此分化、再生的完整植株及其無性系或其后代。在本發明的實施例中,具體使用的是農桿菌介導法。
上述slwrky31p在植物中啟動目的基因表達的應用,也屬于本發明的保護范圍。
上述應用中,所述植物為雙子葉植物,例如番茄、擬南芥或煙草。
擴增slwrky31p的引物對也屬于本發明的保護范圍。
申請人對本發明進行了實際驗證。實驗結果證明,本申請的逆境誘導啟動子slwrky31p在200mmnacl處理24小時的環境下啟動的目的基因的表達量約為正常生長情況下的2.7倍,slwrky31p在300mm甘露醇(mannitol)處理24小時的環境下啟動的目的基因的表達量分別約為正常生長情況下的3.2倍,slwrky31p在1mm水楊酸(salicylicacid,sa)處理24小時的環境下啟動的目的基因的表達量分別約為正常生長情況下的2.9倍。實驗結果表明,本發明的slwrky31p為高鹽、干旱或水楊酸逆境誘導的啟動子,具有較好的效果。
綜上,本發明的slwrky31p可作為構建植物表達載體的元件,將其連接在目的基因之前,使目的基因的表達受高鹽、干旱或水楊酸逆境誘導。因此,本發明的slwrky31p對通過啟動目的基因在高鹽、干旱或水楊酸逆境條件下的高表達,來提高轉基因植物對高鹽、干旱或病原菌的耐受性和抗性,具有重要意義。
附圖說明
本發明將通過例子并參照附圖的方式說明,其中:
圖1為slwrky31p啟動子的pcr擴增電泳圖,其中m為dl2000dnamarker;泳道1為slwrky31p啟動子的pcr擴增條帶。
圖2為將slwrky31p啟動子構建于pbi121sx載體質粒中的示意圖,其中圖a為pbi121sx示意圖,圖b為pbi121sx-slwrky31p示意圖,其中示出了利用slwrky31p啟動子驅動位于其下游的gus基因表達。
圖3為對本發明啟動子進行酶切驗證的結果示意圖,其中m為dl15000dnamarker;泳道1為pbi121sx-slwrky31p重組質粒的電泳條帶;泳道2為pbi121sx-slwrky31p重組質粒經spei和xhoi雙酶切后的電泳條帶。
圖4為轉pbi121sx-slwrky31p的番茄幼苗分別在200mmnacl、300mmmannitol和1mm水楊酸處理24小時環境下幼苗中gus的染色情況。
圖5為轉pbi121sx-slwrky31p的番茄幼苗分別在200mmnacl、300mmmannitol和1mm水楊酸處理24小時環境下幼苗中gus活性的定量統計分析。
具體實施方式
本說明書中公開的所有特征,或公開的所有方法或過程中的步驟,除了互相排斥的特征和/或步驟以外,均可以以任何方式組合。
本說明書中公開的任一特征,除非特別敘述,均可被其他等效或具有類似目的的替代特征加以替換。即,除非特別敘述,每個特征只是一系列等效或類似特征中的一個例子而已。
以下參照具體的實施例來說明本發明。本領域技術人員能夠理解,這些實施例僅用于說明本發現,其不以任何方式限制本發明的范圍。
下述實施例中的實驗方法,如無特殊說明,均為常規方法。下述實施例中所用的材料、試劑等,如無特殊說明,均可從商業途徑得到。
實施例1
(一)逆境誘導啟動子slwrky31p的克隆
1、植物材料
番茄野生型種子為ac+,由四川大學劉永勝教授實驗室惠贈。
2、載體
克隆載體peasy-bluntcloningkit購自北京全式金公司。
3、試劑與藥品
高保真酶primestarhs購自takara公司,瓊脂糖凝膠回收試劑盒購自omegabio-tek公司。引物由生工生物工程(上海)股份有限公司合成,測序由華大基因科技股份有限公司完成。
緩沖液、試劑、細菌培養基配方參見《分子克隆實驗指南》(第三版,作者:著[美]j.莎姆布魯克、黃培堂譯,出版社:科學出版社,isbn:7030103386)。
4、基因克隆
根據sgn(http://solgenomics.wur.nl/)提供的番茄全基因組序列,依據番茄slwrky31基因的上游序列設計擴增引物,正向引物中含有spei識別序列,反向引物中含有xhoi識別序列:
正向:5’-actagtctgggatatatgatgcttattag-3’;
反向:5’-ctcgagtaaaagaaagaagatacaagagtg-3’。
用上述引物以番茄ac+的基因組dna為模板,利用高保真酶primestarhs,擴增slwrky31p啟動子。擴增總體積50μl,其中正向引物1μl,反向引物1μl,dna模板1μl,primestarhs25μl,ddh2o22μl。擴增程序為:98℃10秒,55℃5秒,72℃5秒,30個循環。瓊脂糖凝膠電泳后膠回收與目的啟動子大小相近的片段,連接克隆載體peasy-blunt。將連接產物轉化大腸桿菌,37℃恒溫培養16-18小時。挑取大腸桿菌單克隆,接種含有氨芐青霉素的lb液體培養基中,置于37℃,150轉/每分鐘的搖床中擴大培養16-18小時。將擴大培養的大腸桿菌送華大基因科技股份有限公司進行測序分析,測序正確的克隆用以構建表達載體。
5、膠回收
所用bindingbuffer(xp2)、spwbuffer和elutionbuffer全部來自于omegabio-tek公司的瓊脂糖凝膠回收試劑盒。
(1)在紫外燈下切下含有目的dna的瓊脂糖凝膠,置于1.5ml的離心管中。
(2)在離心管中加入與切下的凝膠塊等體積的bindingbuffer(xp2)。混合均勻后于55-60℃中放置約7分鐘,期間不斷晃動(每2-3分鐘一次),直至凝膠塊完全融化。
(3)將dna吸附管置于2ml離心管中(試劑盒內提供),將上一步完全融化的凝膠液加入到dna吸附管中,8000-10000×g室溫離心1分鐘,棄濾液。
(4)將dna吸附管重新置回2ml離心管中,加入300μl的bindingbuffer(xp2),10000×g室溫離心1分鐘,棄濾液。
(5)將dna吸附管重新置回2ml離心管中,加入700μl的spwbuffer,靜置2-3分鐘,10000×g室溫離心1分鐘,棄濾液。
(6)將dna吸附管重新置回2ml離心管中,10000×g室溫離心1分鐘。
(7)將dna吸附管置于1.5ml離心管中,在吸附管的膜中央加入30-50μl的elutionbuffer,室溫靜置1分鐘。10000×g室溫離心1分鐘洗脫dna片段。
6、與克隆載體的連接
在200μl離心管中依次加入:膠回收dna片段4μl,peasy-bluntcloningvector1μl,輕輕混合,室溫反應10分鐘。反應結束后,將離心管置于冰上,用于大腸桿菌轉化。
7、大腸桿菌轉化
(1)加連接產物于50μltrans1-t1感受態細胞中(peasy-bluntcloning試劑盒提供),輕彈混勻,冰浴20-30分鐘。
(2)42℃熱激30秒,立即置于冰上2分鐘。
(3)加250μl平衡至室溫的soc/lb,200轉每分鐘,37℃孵育1小時。
將菌液于4000轉每分鐘離心1分鐘,棄掉部分上清,保留100-150μl,輕彈懸浮菌體,取全部菌液涂板,37℃培養過夜。
(二)重組表達載體pbi121sx-slwrky31p的構建
1、菌株及載體
大腸桿菌dh5ɑ、根癌農桿菌eha105、表達載體pbi121sx均由西南科技大學生命科學與工程學院生物技術系保存。
2、試劑與藥品
限制性內切酶購自thermo公司,taq酶購自tiangen公司,t4dna連接酶購自promega公司,瓊脂糖凝膠回收試劑盒及小批量質粒提取試劑盒購自omegabio-tek公司。測序由華大基因科技股份有限公司完成。
緩沖液、試劑、細菌培養基配方、大腸桿菌感受態制備參見《分子克隆實驗指南》(第三版,作者:著[美]j.莎姆布魯克、黃培堂譯,出版社:科學出版社,isbn:7030103386)。
3、質粒提取
將測序正確的含有啟動子的peasy-blunt-slwrky31p克隆以及pbi121sx大腸桿菌擴大培養,提取質粒。所用溶液i、溶液ii、溶液iii、bufferhb、washbuffer和elutionbuffer全部來自于omegabio-tek公司的小批量質粒提取試劑盒。
(1)在10-20ml試管中,將攜帶有所需質粒的大腸桿菌接種到5ml含有氨芐青霉素的lb液體培養基,37℃震蕩培養12-16小時。
(2)取1.5-5ml菌液室溫10000×g離心1分鐘。
(3)去上清,加250μl溶液i,渦旋震蕩器震蕩至菌體完全懸浮。
(4)加入250μl溶液ii,溫和顛倒離心管4-6次,獲得澄清的裂解液,室溫孵育2分鐘。
(5)加入350μl溶液iii,溫和顛倒數次混合,至出現白色絮狀沉淀。
(6)室溫13000×g離心10分鐘。
(7)小心吸取上清,移至潔凈的裝配好容積2ml離心管的吸收柱中。室溫10000×g離心1分鐘。
(8)棄濾液,加500μlbufferhb,10000×g離心1分鐘。
(9)棄濾液,用700μlwashbuffer清洗吸收柱,10000×g離心1分鐘。
(10)重復第(9)步。
(11)將吸收柱重新置回2ml離心管中,13000×g離心2分鐘。
將吸收柱放入干凈的1.5ml離心管,在吸附柱的膜中央加30-50μlelutionbuffer,13000×g離心1分鐘。
4、slwrky31p片段和pbi12sx載體的酶切和膠回收
對提取的peasy-blunt-slwrky31p和pbi121sx質粒同時用spei和xhoi進行雙酶切,反應體系如下表1所示,反應條件為37℃酶切2小時。
將酶切后的產物經瓊脂糖凝膠電泳后,分別回收slwrky31p啟動子片段和pbi121sx載體片段。
表1反應體系
5、slwrky31p片段和pbi121sx載體的連接
用t4dna連接酶連接slwrky31p啟動子片段和pbi12sx載體片段,反應體系為:10×t4ligasebuffer1μl,slwrky31p啟動子片段5μl,pbi121sx載體片段3μl,t4ligase1μl。反應條件為:4℃冰箱過夜連接16-18小時。
6、大腸桿菌轉化
將連接產物轉化大腸桿菌。
(1)從超低溫冰箱中取出大腸桿菌感受態細胞,置于冰上解凍。
(2)將連接產物加入到解凍后的感受態細胞中,小心混勻,冰浴30min。
(3)將含有感受態細胞的離心管置于42℃中,熱擊90秒。
(4)快速將熱擊后的離心管轉移至冰浴中,冷卻2-3分鐘。
(5)向冷卻后的離心管中加入800μl的soc,混勻后置于37℃搖床中120轉每分鐘振搖培養1小時。
(6)將菌液于4000轉每分鐘離心1分鐘,棄掉部分上清,保留100-150μl,輕彈懸浮菌體,取全部菌液涂板,37℃培養過夜。
(7)挑取大腸桿菌單克隆進行菌落pcr,將陽性克隆接種含有硫酸卡那霉素的lb液體培養基中,置于37℃,150轉/每分鐘的搖床中擴大培養16-18小時。將擴大培養的大腸桿菌送華大基因科技股份有限公司進行測序分析,測序正確的克隆提取質粒后,用以轉化根癌農桿菌eha105。
7、重組表達載體轉化根癌農桿菌eha105
(1)從超低溫冰箱中取出根癌農桿菌感受態細胞,置于冰上解凍。
(2)將測序正確的重組表達質粒10μl加入到解凍后的感受態細胞中,小心混勻,冰浴30min。
(3)將含有感受態細胞的離心管置于液氮中,速凍1分鐘。
(4)快速將速凍后的離心管轉移至37℃水浴鍋中,放置2-3分鐘。
(5)向離心管中加入800μl的soc,混勻后置于28℃搖床中120轉每分鐘振搖培養4小時。
(6)將菌液于4000轉每分鐘離心1分鐘,棄掉部分上清,保留100-150μl,輕彈懸浮菌體,取全部菌液涂板,28℃培養2天。
(7)挑取農桿菌單克隆進行菌落pcr。
(8)挑取陽性克隆搖菌,并用甘油保存菌液備用。
實施例2利用啟動子slwrky31p驅動報告基因在番茄中表達
1、農桿菌介導的番茄遺傳轉化
將含有pbi121sx-slwrky31p質粒的重組農桿菌轉化番茄,具體方法如下。
(1)將番茄種子滅菌后播種于含有1/2ms固體培養基的培養瓶中,暗培養4d左右,露白后轉置于光照下,培養條件為:25℃,16小時光照,23℃,8小時黑暗,光照強度為80μmolm-2s-1,并在其第一片真葉長出前取下子葉,置于預培養基上培養2d。
(2)將含有pbi121sx-slwrky31p質粒的重組農桿菌接種于5ml含有50μg/ml利福平和50μg/ml硫酸卡那霉素的lb培養基中,28℃搖床培養12小時,得到5mlpbi121sx-slwrky31p種子液。接種1mlpbi121sx-slwrky31p種子液至50ml含有50μg/ml利福平和50μg/ml硫酸卡那霉素的lb培養基中,28℃搖床培養至od600為0.8左右,得到pbi121sx-slwrky31p培養液。將pbi121sx-slwrky31p培養液于室溫4000轉每分鐘,離心4分鐘,棄上清收集菌體,用約10ml誘導培養液重懸菌體,備用。
(3)在滅菌的培養皿中加入40ml的誘導培養液,用移液器吸取800μl重懸菌體加入到含有誘導培養液的培養皿中,混勻。將預培養2天后的番茄子葉浸入誘導培養液和菌體的混合物中10分鐘,期間可用無菌鑷子尖部對子葉進行垂直打孔。
將子葉撈出置于無菌濾紙上吸干液體,隨后將子葉置于共培養基上,于22±1℃、黑暗條件下培養2天后,轉移到分化培養基上;然后,置于光照培養箱中,培養條件為:25℃,16小時光照,23℃,8小時黑暗,光照強度為80μmolm-2s-1。每2周更換一次培養基。當愈傷組織長出2.0cm左右的新芽時,切取嫩芽轉移至生根培養基上生根,培養約4周后獲得完整的硫酸卡那霉素抗性植株。待根系發達后轉移至盆中,在溫室內24±1℃、16小時光照,8小時黑暗條件下培養至番茄果實成熟,并收獲成熟的番茄種子(即t1代轉基因種子)。各培養基成分見表2。
表2番茄組織所用培養基
注:6-ba:6-芐氨基嘌呤(6-benzylaminopurine),iaa:吲哚乙酸(indoleaceticacid),kt:激動素(kinetin),2,4-d:2,4-二氯苯氧基乙酸(2,4-dichlorophenoxyaceticacid)。
(5)將t1代轉基因番茄種子進行滅菌處理得到無菌t1代番茄種子。將無菌t1代番茄種子播種于含50μg/ml硫酸卡那霉素的1/2ms固體選擇培養上,置于光照培養箱中,培養條件為:25℃,16小時光照,23℃,8小時黑暗,光照強度為80μmolm-2s-1。經篩選培養,得到含有硫酸卡那霉素抗性基因的轉基因番茄幼苗。將含有硫酸卡那霉素抗性基因的轉基因番茄幼苗轉移至盆中,在溫室內24±1℃、16小時光照,8小時黑暗條件下培養至番茄果實成熟,并收獲成熟的番茄種子(即t2代轉基因種子)。
2、高鹽、干旱和水楊酸誘導slwrky31p
將t2代轉基因番茄種子進行滅菌處理,得到無菌t2代番茄種子。將無菌t2代番茄種子播種在含有50μg/ml硫酸卡那霉素的1/2ms固體選擇培養。隨后,將兩周大的無菌轉基因番茄幼苗6株置于含有200mmnacl的1/2ms液體培養基中、將兩周大的無菌轉基因番茄幼苗6株置于含有300mmmannitol(甘露醇)的1/2ms液體培養基中、將兩周大的無菌轉基因番茄幼苗6株置于含有1mmsa(水楊酸)的1/2ms液體培養基中、將兩周大的無菌轉基因番茄幼苗6株置于1/2ms液體培養基中,并分別于25℃、16小時光照、23℃、8小時黑暗、平均光照強度為80μmolm-2s-1條件下培養24小時,分別得到200mmnacl處理的番茄幼苗(即鹽脅迫24小時的番茄幼苗)、300mmmannitol處理的番茄幼苗(即干旱脅迫24小時番茄幼苗)、1mmsa處理的番茄幼苗(即水楊酸脅迫24小時番茄幼苗,模擬植物病原菌侵染)、和正常培養24小時的番茄幼苗(作為對照),然后將上述所得的材料進行gus組織化學染色。
3、gus組織化學染色
gus能與顯色底物x-gluc反應,呈現藍色,因而可以通過組織化學染色定性的研究gus的表達水平和表達模式。
(1)gus染色底物的配制:50ml0.5m磷酸緩沖液(ph7.0),50μl100mm鐵氰化鉀k3fe(cn)6(將3.2924g鐵氰化鉀溶于水,定容至100ml,4℃保存),50μl100mm亞鐵氰化鉀k4[fe(cn)6]·3h2o(將4.2239g亞鐵氰化鉀溶于水,定容至100ml,4℃保存),1ml0.5medta,250μl1mg/mlx-gluc(用二甲基甲酰胺溶解,-20℃避光保存)。
(2)染色步驟
染色:將待測樣品浸到gus染液中,37℃保溫箱中放置24-36小時。
脫色:將染色后的樣品經系列濃度乙醇脫色,濃度分別為:50%,75%和90%,至完全脫色后又降低濃度,依次為:90%,70%和50%,最后將樣品保存在50%的乙醇溶液中。
(3)觀察
用體視鏡(leicam205c)分別觀察脫色后的正常培養24小時的番茄幼苗、脫色后的200mmnacl處理24小時的番茄幼苗、脫色后300mmmannitol處理24小時的番茄幼苗和脫色后1mmsa處理24小時的番茄幼苗的并拍照,結果見圖4。結果顯示,正常培養24小時的番茄幼苗經gus染色后,僅在番茄幼苗的根和葉片中的少數的部位呈現出微弱的淺藍色,而在200mmnacl處理24小時或300mmmannitol處理24小時的的番茄幼苗的根、莖和葉片也都呈現深藍色,并且在1mmsa處理24小時的番茄幼苗的葉片也都呈現深藍色。gus染色實驗結果表明,相對正常培養的番茄,300mmmannitol或200mmnacl處理的番茄gus表達量在根、莖和葉片中都顯著增加,而1mmsa處理番茄gus表達量在葉片中顯著增加,在莖和根中也略有增加。
4、gus酶活的測定
按照cote等的方法(cotec,rutledgerg.animprovedmugfluorescentassayforthedeterminationofthegusactivitywithintransgenictissueofwoodyplants.plantcellreport,2003,21(6):619-624)對正常培養24小時的番茄幼苗、200mmnacl處理24小時的番茄幼苗、300mmmannitol處理24小時的番茄幼苗和1mmsa處理24小時的番茄幼苗進行gus活性的熒光定量分析,實驗結果如圖5所示,實驗結果顯示:正常培養24小時的番茄的gus活性值為0.96nmol4-mumin-1mg-1蛋白,200mmnacl處理24小時的番茄的gus活性值為2.55nmol4-mumin-1mg-1蛋白,300mmmannitol處理24小時的番茄的gus活性值為3.06nmol4-mumin-1mg-1蛋白,1mmsa處理24小時的番茄的gus活性值為2.73nmol4-mumin-1mg-1蛋白,nacl處理、mannitol處理和sa處理24小時下gus活性分別是正常培養的2.7倍、3.2倍和2.9倍,呈極顯著差異。上述結果表明啟動子slwrky31p受高鹽、干旱和sa的誘導。
在植物的免疫應答中,水楊酸(sa)被認為是一種十分重要的病源信號分子,植物體感染病原菌后體內sa會急劇增加。所以本實施例采用sa處理來模擬病原菌對植物的侵染。
本發明并不局限于前述的具體實施方式。本發明擴展到任何在本說明書中披露的新特征或任何新的組合,以及披露的任一新的方法或過程的步驟或任何新的組合。
序列表
<110>西南科技大學
<120>一個逆境誘導啟動子slwrky31p的克隆及其應用
<130>2017
<160>3
<170>patentinversion3.3
<210>1
<211>1849
<212>dna
<213>番茄(solanumlycopersicum)
<400>1
ctgggatatatgatgcttattagagttgcacataatgtgcttcatgtagtgtacttaacc60
tcttaatagagtgcattacatgaagcactattatagtgatacaatccaacttgttctgca120
catactcaattagctctattgtttatctttcgaaaaaaaaacccgatctaaattgatata180
ttgattagatgtgccatttaattaattgagaatacaatatgtgatttctcagataaaatg240
gaagtcaaaaattcttctttggaaatagtttttcttttaacaataataataataataata300
atcaaaagttatccaaatggctcaaaataattacttcaagtaagtaaaagaaattaagat360
tacattattaaacttttcattaattacgtaccatgatatttaatttccagcttctttgat420
ggcatctacaagcttgagttgaaggtggctacgaatacggacgccgaagatggcacagat480
ttcaatgtccgccaggcttcgagcgaaaaatttcacgatccactgagattcagtttttta540
tcattaaaaaataaaaaaaaaacaaaaaagaaagcgaaaacctgctgcattggttaaaca600
cagcggccagtaaactgatcaaatcttttattttggccacaatcatttaaatatataatt660
gctgaaagaagaatatacacctctagaaagaaactttttcctgttccaatcgttatccat720
tttaagtgtataaacaattaaatgaataaaataacgggaaatatccattagagagcatga780
catttgttatttgtccaataaaatttgatgtgcttgcttctttgacttgtatacatttag840
cttaaattgactttatacttaaacaaatgaatttctctttaattaataatcagagtaacg900
taatcctaaaattctagtatattaatcataaaacatataataaaaacacattgtaaaata960
gcacttgcaacatatctcgactatctagactgatatcgataatatattctgagaaacttc1020
ttttgtgatcatttattacttattaggttaaagataaaaactcctttgttttgatgcaaa1080
tagaataatacacaaccacctttaactctcactaatgagcataaatgattaaaatgttta1140
aaacaaaattgaattgtatcattagaaaattaaaaaaagaaaaaagttaattcatttttt1200
tattgctataaatatttagttttaaatctatccaaacaaggttcgaaaagaatggaaaaa1260
gattagataaaaaggatattgacaattcgcccgataaaagagcctcgtgattgattgatt1320
gttagttaatcaactgatagtggccagtggggtaccaccatcacaacgacagcgacataa1380
cgcgtttggcgtaattgcattacaatagcagcctctcttttcttgtacaatttgtgtaaa1440
tgtatctattatagtcaaagccaccaattttgactcaacacacaaatagtattatttttt1500
agatttcattttccacggtcaaagctattttgaggatcttagaagtttctttaaatatct1560
atagaatagaaaaataaaaaagacaatgagcttggatgacactgttttattttttaatta1620
aaaactttagaatttatatttgacagaaaggaaattttattaaaaatgtcaattatcaaa1680
taaattattaaatttaatcaaattttaatattatcgagtgagaaatgaaaaaaaaaaagc1740
tagtggattgtagaatacgtgttcatttttgtttttaatatcccacccactccaaacgaa1800
aggttatataatcgaattcccattgcactcttgtatcttctttctttta1849
<210>2
<211>29
<212>dna
<213>人工序列
<400>2
actagtctgggatatatgatgcttattag29
<210>3
<211>30
<212>dna
<213>人工序列
<400>3
ctcgagtaaaagaaagaagatacaagagtg30
- 還沒有人留言評論。精彩留言會獲得點贊!
- 大豆低溫誘導型啟動子、包含該啟動子的重組表達載體及應用的制作方法與工藝
- 受植物激素調控的啟動子及其應用的制作方法與工藝
- 一種受鹽誘導的特異性誘導型啟動子DNA序列及應用的制作方法與工藝
- 受維甲酸調控的具有濃度依賴效應的重組啟動子及其使用的制作方法與工藝
- 一種磷酸鹽誘導啟動子降低黑曲霉β?葡萄糖苷酶表達酶活的方法與流程
- 一種疫霉誘導性人工合成啟動子PMP1及其重組表達載體和應用的制作方法與工藝
- 一種植物鹽/干旱誘導型啟動子soyEQ及其應用的制作方法與工藝
- 新型的L-蘇氨酸-誘導型啟動子及其應用的制作方法與工藝
- 一種植物茉莉酸甲酯誘導表達啟動子PosMeJ1及其應用的制作方法與工藝
- 一個逆境誘導啟動子SlWRKY31P的克隆及應用的制造方法與工藝