一種黃曲霉素B1分子印跡聚合物的制備方法與流程
本發明涉及一種分子印跡聚合物的制備方法,特別是一種黃曲霉素b1分子印跡聚合物的制備方法。
背景技術:
黃曲霉毒素(aft)在1993年就已經被世界衛生組織(who)的癌癥研究機構劃定為i類致癌物。在天然污染的食品和飼料中以黃曲霉毒素b1(afb1)最為常見,其毒性和致癌性也最強,不僅給社會帶來重大的經濟損失,而且嚴重威脅消費者的健康。世界各國及地區均制定了嚴格的aft限量標準,且限量要求日益嚴格。目前黃曲霉毒素的檢測方法有薄層層析法、高效液相色譜法、酶聯免疫吸附法、放射免疫測定法、免疫親和層析柱-高效液相色譜法、免疫親和層析柱-熒光光度法等。目前免疫親和層析柱-高效液相色譜法和免疫親和層析柱-熒光光度法,具有快速、靈敏、準確的特點,已被國家標準gb/t18979-2003所采用。然而,免疫親和柱所需的抗體制備不易,且質量穩定性與產品穩定性都難以控制。
分子印跡聚合物是一類以小分子化合物為模板,通過物理包埋和化學聚合而制備的新型功能材料。分子印跡的制備理念與抗原抗體的親和原理非常接近。由于分子印跡聚合物可選擇的聚合單體具有化學多樣性、能夠充分預測最終聚合物的結構和功能屬性,該聚合物通常都選擇結構上沒有活潑基團的單體,因此具有熱穩定性好和使用壽命長等優點。由于其聚合過程中包埋了分子印跡,而聚合物的化學穩定性導致分子印跡能夠非常穩定的保持下去。因此,分子印跡聚合物可以作為高靈敏度的物理吸附劑廣泛應用于生物、藥品和食品等樣品處理及檢測等方面。
殼聚糖作為一種廉價易得的生物高分子材料已經被應用于分子印跡的研究之中。殼聚糖分子中具有氨基和羥基,因此改性的方式多,反應靈活。然而,如何利用殼聚糖上的活性基團,制備出特異性的分子印跡是一個研究難點。
技術實現要素:
本發明的目的是提供一種黃曲霉素b1分子印跡聚合物的制備方法,為現有黃曲霉素b1檢測方法提供了全新的思路,利用針對黃曲霉素b1的分子印跡聚合物作為吸附劑,對樣品中的黃曲霉素b1的含量進行檢測,具有操作方便,聚合物可做到無限次回收套用,對于黃曲霉素b1的檢測靈敏度高,檢測結果準確等優點。
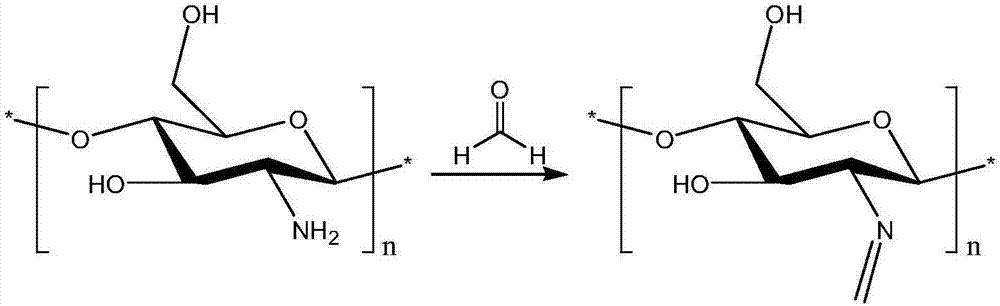
為實現上述目的,本發明提供的黃曲霉素b1分子印跡聚合物的制備方法,利用單醛對殼聚糖進行預交聯,所得的預交聯殼聚糖與甲基環氧氯丙烷反應得到殼聚糖環氧樹脂預聚物;殼聚糖環氧樹脂預聚物與黃曲霉素b1混合后,進行聚合;然后通過對包埋了黃曲霉素b1的殼聚糖環氧樹脂進行物理、化學清洗,最終得到殼聚糖環氧樹脂,用殼聚糖環氧樹脂即可快速檢測出黃曲霉素b1。
本發明利用親水性殼聚糖作為聚合單體,首先用單醛封閉殼聚糖上的氨基反應位點,能夠完美的控制殼聚糖中氨基的活性,從而使與甲基環氧氯丙烷的反應位點控制在殼聚糖上的羥基位置,同時也保證了殼聚糖環氧樹脂與黃曲霉素b1不會發生共價結合;單醛上的烷基鏈以及環氧基的聚合反應發生后產生的結合態氧為黃曲霉素b1提供了良好的極性與非極性環境。
本發明提供的一種黃曲霉素b1分子印跡聚合物的制備方法,具體的制備步驟如下:
1)殼聚糖預交聯步驟如下:
將一定量的殼聚糖溶于乙酸水溶液,置于裝有攪拌器及溫度計的500ml三口燒瓶中;加入液體石蠟,啟動攪拌器,常溫下攪拌10min后升溫到50℃,滴加適量的乳化劑,乳化10min,然后升溫到60℃,滴加一定量的單醛,攪拌反應3h;冷卻,加入水溶性溶劑幫助沉淀,過濾,濾餅用不溶于水的溶劑洗滌三次,將過濾得到的濾餅于60℃真空干燥,得到殼聚糖預交聯產物。
其中,所述的殼聚糖交聯所用的乳化劑為span60、吐溫80、卵磷脂、大豆磷脂中的一種或幾種,優選為span60、吐溫80或卵磷脂;所述的殼聚糖交聯所用的單醛為碳鏈長度1-5的單醛,優選為甲醛、乙醛或丙醛;所述水溶性溶劑為碳鏈長1-5的短鏈醇、丙酮,優選為甲醇、乙醇或丙酮;所述的不溶于水的溶劑為乙醚、沸點30-60的石油醚、沸點60-90的石油醚、正己烷、環己烷,優選為石油醚、正己烷或環己烷。
2)殼聚糖環氧樹脂預聚物步驟如下:
將步驟1)得到的殼聚糖預交聯產物分散于適量活化劑中,升溫到90℃,反應4h,反應完畢,利用旋轉蒸發儀將反應體系的溶劑蒸干,將蒸干剩余物溶于不溶于水的溶劑中,以恒壓滴液漏斗緩慢滴加質量分數為48.5%的氫氧化鈉水溶液,反應4h,反應完畢用水洗至中性,得到殼聚糖環氧樹脂預聚物混合液。
其中,所述的活化劑為甲基環氧氯丙烷;所述的不溶于水的溶劑為正己烷、環己烷、苯、甲苯、乙苯,優選為甲苯、乙苯或正己烷。
3)黃曲霉素b1分子印跡聚合物步驟如下:
取黃曲霉素b1加入步驟2)得到的殼聚糖環氧樹脂預聚物混合液中,0℃條件下以恒壓滴液漏斗緩慢滴加固化劑,反應2h,將反應液分散于不溶于水的溶劑中,得到的固體沉降,過濾,用溶于水的溶劑洗滌三次,干燥,最終得到黃曲霉素b1分子印跡聚合物。
其中,所述的固化劑為乙二胺、己二胺、二乙烯三胺、三乙烯四胺、二乙氨基丙胺、順丁烯二酸酐、鄰苯二甲酸酐,三氟化硼乙醚溶液(cas:109-63-7,市售);優選為三氟化硼乙醚溶液、己二胺或順丁烯二酸酐;所述的不溶于水的溶劑為二氯甲烷、二氯乙烷、氯仿、乙酸乙酯、正己烷、環己烷,優選為二氯甲烷、正己烷或二氯乙烷;所述的溶于水的溶劑為碳鏈長1-5的短鏈醇、丙酮,優選為甲醇、乙醇或丙醇。
本發明的優點和有益效果在于:
(1)本發明提供的黃曲霉素b1分子印跡聚合物的制備方法,利用親水性殼聚糖作為聚合單體。a)首先用單醛封閉殼聚糖上的氨基反應位點,保證了殼聚糖環氧樹脂預聚物的順利合成;b)用單醛封閉殼聚糖上的氨基反應位點,保證了殼聚糖環氧樹脂與黃曲霉素b1不會發生共價結合;c)單醛上的烷基鏈為黃曲霉素b1中的疏水基團提供了完美的疏水環境,使得殼聚糖環氧樹脂預聚物與黃曲霉素b1具有良好的親附性;d)環氧基的聚合反應發生后產生的結合態氧為黃曲霉素b1提供了良好的極性環境。
(2)采用本發明提供的方法制備黃曲霉素b1分子印跡聚合物,可快速檢測出黃曲霉素b1,且黃曲霉素b1的回收率高達98.5%,該分子印跡化合物使用壽命長,經過脫附洗滌可重復利用。
附圖說明
圖1為實施例1殼聚糖的預交聯化學反應圖。
圖2為實施例1殼聚糖環氧樹脂預聚物化學反應圖。
圖3為黃曲霉素b1分子印跡聚合物化學反應示意圖。
圖4為殼聚糖、實施例1、實施例2、實施例3化合物的紅外譜圖,其中a為殼聚糖、b為實施例1的分子印跡聚合物、c為實施例2的分子印跡聚合物、d為實施例3的分子印跡聚合物。
具體實施方式
為使本領域技術人員清楚明白本發明的技術方案,下面通過以下實施例進一步說明本發明的內容,但不應理解為對本發明的限制。
實施例1
1)殼聚糖的預交聯:
將2.02g殼聚糖溶于200ml2%乙酸水溶液,置于裝有攪拌器及溫度計的500ml三口燒瓶中。加入200ml液體石蠟,啟動攪拌器,常溫下攪拌10min后升溫到50℃,滴加3mlspan60,乳化10min,然后升溫到60℃,滴加5.8g的甲醛,攪拌反應3h。冷卻,加入200ml丙酮幫助沉淀,過濾,濾餅用100ml石油醚洗滌三次,將過濾得到的濾餅于60℃真空干燥,得到殼聚糖預交聯產物。
2)殼聚糖環氧樹脂預聚物的制備:
將步驟1)得到的殼聚糖預交聯產物分散于30ml甲基環氧氯丙烷中,升溫到90℃,反應4h,反應完畢,利用旋轉蒸發儀將反應體系的溶劑蒸干,將蒸干剩余物溶于50ml甲苯,以恒壓滴液漏斗緩慢滴加2.8g質量分數為48.5%的氫氧化鈉水溶液,反應4h,反應完畢用水洗至中性,得到殼聚糖環氧樹脂預聚物混合液。
3)黃曲霉素b1分子印跡聚合物的制備:
取1.8g黃曲霉素b1加入步驟2)得到的殼聚糖環氧樹脂預聚物混合液中。0℃條件下以恒壓滴液漏斗緩慢滴加1.5ml三氟化硼/乙醚溶液,反應2h,將反應液分散于100ml二氯甲烷中,得到的固體沉降,過濾,用100ml乙醇洗滌三次,干燥,最終得到黃曲霉素b1分子印跡聚合物。
實施例2
1)殼聚糖的預交聯:
將2.02g殼聚糖溶于200ml2%乙酸水溶液,置于裝有攪拌器及溫度計的500ml三口燒瓶中。加入200ml液體石蠟,啟動攪拌器,常溫下攪拌10min后升溫到50℃,滴加3ml吐溫80,乳化10min,然后升溫到60℃,滴加7.5g的乙醛,攪拌反應3h。冷卻,加入200ml甲醇幫助沉淀,過濾,濾餅用100ml正己烷洗滌三次,將過濾得到的濾餅于60℃真空干燥,得到殼聚糖預交聯產物。
2)殼聚糖環氧樹脂預聚物的制備:
將步驟1)得到的殼聚糖預交聯產物分散于30ml甲基環氧氯丙烷中,升溫到90℃,反應4h,反應完畢,利用旋轉蒸發儀將反應體系的溶劑蒸干,將蒸干剩余物溶于50ml乙苯,以恒壓滴液漏斗緩慢滴加2.9g質量分數為48.5%的氫氧化鈉水溶液,反應4h,反應完畢用水洗至中性,得到殼聚糖環氧樹脂預聚物混合液。
3)黃曲霉素b1分子印跡聚合物的制備:
取1.8g黃曲霉素b1加入步驟2)得到的殼聚糖環氧樹脂預聚物混合液中。0℃條件下以恒壓滴液漏斗緩慢滴加1.5ml乙二胺,反應2h,將反應液分散于100ml正己烷中,得到的固體沉降,過濾,用100ml甲醇洗滌三次,干燥,最終得到黃曲霉素b1分子印跡聚合物。
實施例3
1)殼聚糖的預交聯:
將2.02g殼聚糖溶于200ml2%乙酸水溶液,置于裝有攪拌器及溫度計的500ml三口燒瓶中。加入200ml液體石蠟,啟動攪拌器,常溫下攪拌10min后升溫到50℃,加入3.5g卵磷脂,乳化10min,然后升溫到60℃,滴加9.4g的丙醛,攪拌反應3h。冷卻,加入200ml乙醇幫助沉淀,過濾,濾餅用100ml環己烷洗滌三次,將過濾得到的濾餅于60℃真空干燥,得到殼聚糖預交聯產物。
2)殼聚糖環氧樹脂預聚物的制備:
將步驟1)得到的殼聚糖預交聯產物分散于30ml甲基環氧氯丙烷中,升溫到90℃,反應4h,反應完畢,利用旋轉蒸發儀將反應體系的溶劑蒸干,將蒸干剩余物溶于50ml正己烷,以恒壓滴液漏斗緩慢滴加3.1g質量分數為48.5%的氫氧化鈉水溶液,反應4h,反應完畢用水洗至中性,得到殼聚糖環氧樹脂預聚物混合液。
3)黃曲霉素b1分子印跡聚合物的制備:
取1.8g黃曲霉素b1加入步驟2)得到的殼聚糖環氧樹脂預聚物混合液中。0℃條件下以恒壓滴液漏斗緩慢滴加1.7ml順丁烯二酸酐,反應2h,將反應液分散于100ml二氯乙烷中,得到的固體沉降,過濾,用100ml丙醇洗滌三次,干燥,最終得到黃曲霉素b1分子印跡聚合物。
實施例4分子印跡聚合物中黃曲霉素b1負載量計算
取上述實施例制備的三種微球,分別加入到黃曲霉素b1的甲醇溶液中,在搖床中振蕩吸附,在吸附時間內分時間點取上清液測定含量。待吸附飽和,測定溶液中黃曲霉素b1的濃度變化,利用下列公式來計算靜態吸附量。實施例1至實施例3分子印跡聚合物靜態吸附數據見表1。
q0=(co-cx)*v0/mx
q0:靜態吸附量(mg/g);
v0:黃曲霉素b1溶液體積(ml);
co:黃曲霉素b1溶液的初始濃度(mg/ml);
cx:吸附平衡后黃曲霉素b1溶液的濃度(mg/ml);
mx:吸附平衡時微球的用量(g)。
實施例5分子印跡聚合物中黃曲霉素b1的回收率計算
取吸附飽和后三種分子印跡聚合物先經布氏漏斗抽濾,然后在漏斗上方緩慢加入少量二次水洗滌,然后將微球放入索氏提取器中,以甲醇作為萃取劑對微球中吸附的物質進行循環萃取,分時間點取萃取液測定含量,直至萃取液的濃度不發生變化,利用下式確定黃曲霉素b1的回收率。回收率數據見表1。
uy=(cx*v1/q0*mx)*100%
uy:靜態脫附率(%);
cx:回收完成后溶液中黃曲霉素b1的濃度(mg/ml);
v1:回收溶劑總體積(ml);
mx:吸附平衡時微球的用量(g)。
表1:分子印跡聚合物對黃曲霉素b1的吸附量以及回收率數據
參閱圖4,從未改性的殼聚糖的紅外光譜可以看到,3200cm-1~3500cm-1處具有一個較大的寬峰,從歸屬上來看,應為o-h的伸縮振動吸收峰存在的區間以及n-h的伸縮振動吸收峰存在的區間,因此考慮是兩種伸縮振動吸收峰重疊而成。而1000cm-1附近的吸收峰則可以歸屬為殼聚糖分子中所具有的羥基中的c-o伸展與o-h面內變形振動吸收峰的這兩個峰疊加到一起的結果。而1601cm-1處的吸收峰則是n-h的彎曲振動吸收帶所提供的。
而三種分子印跡聚合物的紅外光譜表明:1)氨基的振動吸收峰(3358cm-1處的小分叉)消失表明其發生了化學反應;2)氨基的彎曲振動吸收帶消失,也證明了這一點;3)飽和c-h伸縮振動的吸收峰2924cm-1和2869cm-1比未改性的殼聚糖明顯增強,這說明希夫堿的形成使得殼聚糖上引入了碳鏈。
- 還沒有人留言評論。精彩留言會獲得點贊!