固態紅色硅烷化碳點及其制備方法與流程
本發明屬于熒光納米材料合成技術領域,具體涉及一種固態紅色硅烷化碳點(CDs)及其制備方法。
背景技術:
熒光碳納米材料具有較好的熒光性能與低毒性,在信息安全、光催化藥物傳輸、細胞成像、傳感、光電器件以及疾病診斷等領域具有良好的應用前景。作為熒光碳納米材料之一的碳點因其獨特的光學性質和環境友好性,從而受到人們的廣泛關注。
但目前固態碳量子點難以制得,因為當碳點處于聚集態時,極易引起熒光猝滅。到目前為止,本領域的絕大部分工作都是關于碳點水溶液的熒光性能與應用,很少有報道涉及到碳點雜化固相材料和碳點團聚體的固態發光性能及相關應用。
本領域關于碳點發光性能的研究,絕大部分限于短波長的熒光(藍色、綠色),而長波長的熒光(橙光、紅光)報道很少。而長波長的熒光在特殊應用中有著短波長熒光無法企及的優良特性,所以對于紅色熒光碳點的研究顯得尤為重要。雖然目前已經有關于紅光碳點的報道,但均是表現為液態熒光,對于固態紅色熒光碳點的研究顯得尤為重要。
為了克服現有技術的缺點與不足,本發明提供了一種新型固態紅色碳點及其制備方法。本發明的制備方法操作工藝簡單、快捷、原料廉價易得、產物穩定性好(如圖9、10),不潮解,對于碳點的制備過程簡單,后處理容易,粒子大小均勻(如圖3所示),熒光效果顯著,所述碳點不具備典型的熒光激發依賴性質(如圖8所示),不同于絕大多數文獻所報道的具有的激發依賴性質的碳點。
技術實現要素:
本發明公開了一種固態紅色硅烷化碳點及其制備方法與應用,所述方法為回流法。本發明通過簡易快捷的回流反應,無需后續強酸或表面鈍化劑的處理,即可一步快速獲得固態碳點,原料簡單易得,價格低廉,反應條件溫和。解決了現有固態紅色碳點因制備工藝和原料的限制而無法規模化生產的問題,制備得到的碳點穩定性好(如圖9、10),不潮解,粒子大小均勻(如圖3所示),熒光效果顯著,所述碳點不具備典型的熒光激發依賴性質(如圖8所示),不同于絕大多數文獻所報道的具有的激發依賴性質的碳點。
一方面,本發明提供了一種新型固態紅色碳點的制備方法。本發明的制備方法操作工藝簡單、快捷、原料廉價易得、產物穩定性好。
本發明提供的本發明所述碳點的制備方法,所述方法對于碳點的制備過程簡單,后處理容易,粒子大小均勻(如圖3所示),穩定性好(如圖9、10),熒光效果顯著,所述碳點不具備典型的熒光激發依賴性質(如圖8所示),激發改變,發射峰位不變,其不同于絕大多數文獻所報道的具有的激發依賴性質的碳點。
一方面,本發明涉及一種固態紅色硅烷化碳點的制備方法,其中,所述方法為回流法,包括以下步驟:將檸檬酸溶解于丙酮中得到混勻的溶液,然后將所述混勻的溶液加至硅烷偶聯劑中,加熱反應,得到粘稠態的碳點,再將所得粘稠態的碳點放置于烘箱中烘干得到固態碳點。
在一些實施方案,本發明所述的方法,其中,將所得粘稠態的碳點放置于60℃烘箱中24h得到固態碳點。
在一些實施方案,本發明所述的方法,其中,所述加熱反應是在150-240℃下進行的。
在一些實施方案,本發明所述的方法,其中,所述加熱反應的反應時間為5-60min。
在一些實施方案,本發明所述的方法,其中,所述檸檬酸、丙酮和硅烷偶聯劑的用量比為1~4g:5~10mL:10~20mL。
在一些實施方案,本發明所述的方法,其中,所述檸檬酸、丙酮和硅烷偶聯劑的用量比為1g:5mL:10mL。
在一些實施方案,本發明所述的方法,其中,所述硅烷偶聯劑包括含有2-3個烷氧硅基的氨基硅烷偶聯劑、亞氨基硅烷偶聯劑、環氧基硅烷偶聯劑和巰基硅烷偶聯劑中的至少一種。
在一些實施方案,本發明所述的方法,其中,所述硅烷偶聯劑包括N-(β-氨乙基)-γ-氨丙基-甲基二甲氧基硅烷、N-氨乙基-γ-氨丙基-三甲氧基硅烷、3-氨丙基三乙氧基硅烷、甲基三乙氧基硅烷和3-疏醇基丙基三甲氧基硅烷中的至少一種。
在一些實施方案,本發明所述的方法,其中,所述硅烷偶聯劑為N-(β-氨乙基)-γ-氨丙基-甲基二甲氧基硅烷。
另一方面,本發明涉及一種本發明所述的方法制備得到的固態紅色硅烷化碳點,其中所述碳點粒徑在5-7nm,其透射電鏡圖基本上如圖3所示。
在一些實施方案,本發明所述的碳點具有一定程度的石墨化結構,其高分辨透射電鏡圖基本上如圖4所示。
在一些實施方案,本發明所述的碳點含有Si元素,且C元素含量達到約63.42%,其XPS譜圖基本上如圖5所示。
在一些實施方案,本發明所述的碳點的XRD譜圖基本上如圖6所示。
本發明的目的通過下述技術方案實現:硅烷化紅色固態碳點,包括最優制備條件和后處理方法。
一種固態紅色硅烷化碳點的制備方法,包括以下步驟:
所述碳點的制備方法為回流法,包括以下步驟:
(1)將檸檬酸溶解于丙酮中,超聲分散使其完全溶解,得到溶液;
(2)將上述步驟(1)中得到的溶液加入到硅烷偶聯劑中,進行回流反應得到流動粘稠態的紅色碳點;
(3)將步驟(2)中得到的流動粘稠態碳點置于60攝氏度烘箱中靜置24小時,得到固態紅色碳點;
所述的檸檬酸、丙酮和硅烷偶聯劑的用量比為1g:5mL:10mL。
所述的回流反應優選為在150-240℃下反應5-60min,更優選為在150℃下回流5min。
本發明中所述的硅烷偶聯劑包括但不限于含有2-3個烷氧硅基的氨基硅烷偶聯劑、亞氨基硅烷偶聯劑、環氧基硅烷偶聯劑和巰基硅烷偶聯劑中的至少一種,如N-(β-氨乙基)-γ-氨丙基-甲基二甲氧基硅烷(AEAPMS),N-氨乙基-γ-氨丙基-三甲氧基硅烷(AEATMS),3-氨丙基三乙氧基硅烷(APTES),甲基三乙氧基硅烷(METS),3-疏醇基丙基三甲氧基硅烷(MPTES)等。更優選為N-(β-氨乙基)-γ-氨丙基-甲基二甲氧基硅烷(AEAPMS)。
(4)步驟(3)得到的碳點穩定性好,在紫外光下可以發出明亮的紅光,可與多種物質進行復合,得到性能優異的復合材料。
本發明使用的術語“基本上如圖所示”是指透射電鏡圖、XPS譜圖或XRD譜圖中至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或至少95%,或至少99%的特征或峰顯示在其圖中。
在本發明的上下文中,當使用或者無論是否使用“大約”或“約”等字眼時,表示在給定的值或范圍的10%以內,適當地在5%以內,特別是在1%以內。或者,對于本領域普通技術人員而言,術語“大約”或“約”表示在平均值的可接受的標準誤差范圍內。每當公開一個具有N值的數字時,任何具有N+/-1%,N+/-2%,N+/-3%,N+/-5%,N+/-7%,N+/-8%或N+/-10%值以內的數字會被明確地公開,其中“+/-”是指加或減。
本發明的原理:
本發明利用檸檬酸,丙酮和硅烷偶聯劑回流反應,原料簡單易得,價格低廉,反應條件溫和,制備的碳點產量高,熒光性能穩定,可以很好的應用于光電裝置,光催化,生物成像及農用等領域。
本發明相對于現有技術,具有如下的優點及效果:
(1)本發明使用檸檬酸,丙酮和硅烷偶聯劑回流反應,無需后續強酸或表面鈍化劑處理,即可簡單快速獲得固態碳點。
(2)本發明原料廣泛易得,價格低廉,制備條件溫和,低能耗僅需150℃即可反應,且產率高,可以制得8mL液態粘稠態碳點,有望大規模工業化生產。
(3)本發明制得的碳點穩定性好,不潮解,且光穩定好(如圖9、10),粒子大小均勻(如圖3所示),所述碳點不具備典型的熒光激發依賴性質(如圖8所示),激發改變,發射峰位不變,其不同于絕大多數文獻所報道的具有的激發依賴性質的碳點,可應用于光電、光催化、氣體傳感以及農用等領域。
附圖說明
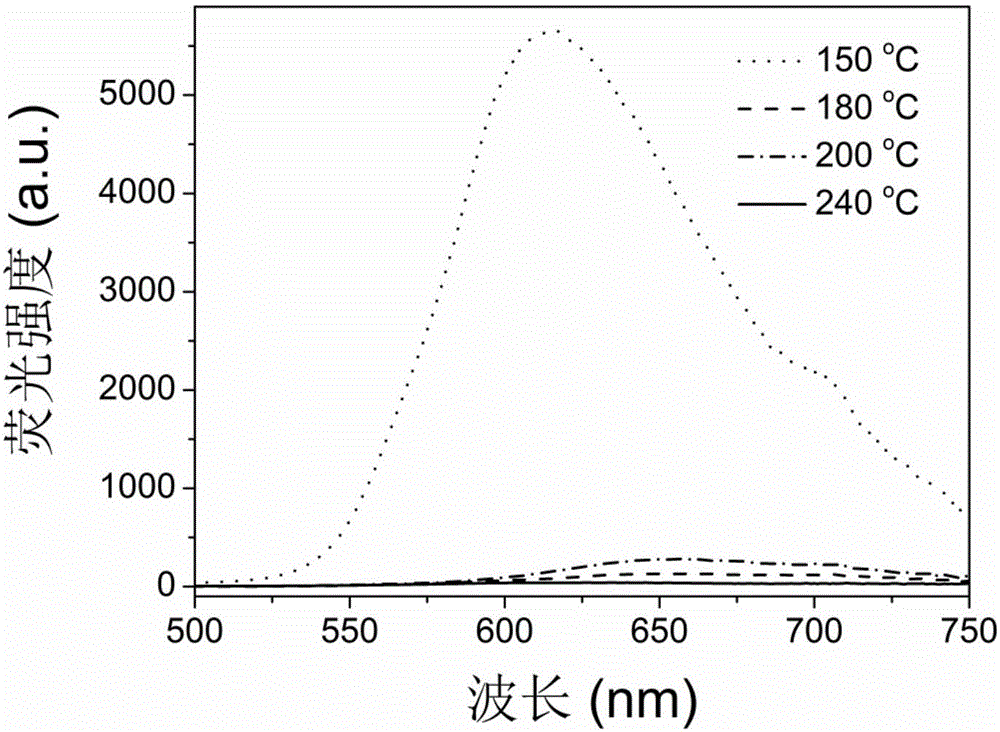
圖1是實施例1在不同反應溫度下制得的碳點的發射光譜圖(λex=365nm)。
圖2是實施例2在不同反應時間下制得的碳點的發射光譜圖(λex=365nm)。
圖3是實施例3在最優反應溫度(150℃)與最優反應時間(5min)下制得的碳點的透射電鏡圖。
圖4是實施例3在最優反應溫度(150℃)與最優反應時間(5min)下制得的碳點的高分辨透射電鏡圖。
圖5是實施例3制得的碳點的XPS譜圖。
圖6是實施例3制得的碳點的XRD譜圖。
圖7是實施例3制得的碳點的紅外譜圖。
圖8是實施例3制得的碳點在不同激發下的發射光譜圖。
圖9是碳點在紫外燈下的穩定性研究結果。
圖10是碳點自然光下的穩定性研究。
具體實施方式
下面結合實施例及附圖對本發明作進一步詳細的描述,但本發明的實施方式不限于此。
實施例1
1g檸檬酸溶于5mL丙酮中,超聲使其完全溶解,再將得到的溶液加入到10mL的硅烷偶聯劑(AEAPMS,工業級別,購于廣州龍凱化工有限責任公司)中,在150-240℃下進行回流反應10min。即可制得在不同反應溫度下的碳點,其發射光譜圖如圖1所示(用天美(中國)科學儀器有限公司的熒光光譜儀F-7000進行測試得到)。
實施例2
1g檸檬酸溶于5mL丙酮中,超聲使其完全溶解,再將得到的溶液加入到10mL的硅烷偶聯劑(AEAPMS,工業級別,購于廣州龍凱化工有限責任公司)中,在150℃下進行回流反應5-60min。即可制得在不同反應時間下的碳點,其發射光譜圖如圖2所示(用天美(中國)科學儀器有限公司的熒光光譜儀F-7000進行測試得到)。
實施例3
1g檸檬酸溶于5mL丙酮中,超聲使其完全溶解,再將得到的溶液加入到10mL的硅烷偶聯劑(AEAPMS,工業級別,購于廣州龍凱化工有限責任公司)中,在150℃下進行回流反應5min(即最優反應溫度與反應時間),然后冷卻至室溫即可制得呈現粘稠態的碳點。對制得的碳點進行觀察與測試,結果見圖3-8。
參見圖3,是本實施制備的碳點的透射電鏡圖,從圖中可以明顯看出碳點是均分分布的,分散性良好,粒徑在5-7nm。
參見圖4,是本實施制備的碳點的高分辨透射電鏡圖,從圖中可以明顯看出碳點是具有明顯的晶格條紋,說明制得的碳點具有一定程度的石墨化結構。
參加圖5,是本實施制得的碳點的XPS譜圖及其元素含量,從圖中可以明顯看出材料中含有Si元素的存在。
參見圖6,是本實施制得的碳點的XRD譜圖及其元素含量,從圖中可以明顯看出碳點呈現寬峰,峰位在25度,這與碳點的石墨化結構的特征峰對應,對應為石墨的[002]晶面,這可以更進一步解釋圖4的石墨化的晶格條紋。這也進一步證實了該材料為石墨化了的碳點。
參見圖7,是本實施制得的碳點的紅外光譜圖,從圖中可以看出該碳點中含有Si-O-Si、Si-O-CH、Si-CH2鍵的吸收,表明了制得的產物是硅烷功能化的碳點。
參加圖8,是本實施制得的碳點在不同激發波長下的發射光譜圖,從圖中可以看出,隨著激發波長的持續增加500-580,熒光強度持續增加,峰位在640nm,表面所得碳點為明顯的紅光發射,所述碳點不具備典型的熒光激發依賴性質,不同于絕大多數文獻所報道的具有的激發依賴性質的碳點。
上述實施例中的硅烷偶聯劑除了AEAPMS外,還可以使用APTES、AEATMS或MPTES等其他硅烷偶聯劑代替,其效果一致。
本發明還詳細探討了本發明所述碳點的穩定性:
1碳點在紫外燈下的穩定性研究
取2mL的粘稠態碳點于石英比色皿中,將光譜儀的雙狹縫調為5,電壓調為500V,激發波長與發射波長分別設為360nm和440nm,使其持續激發,研究紫外光下碳點熒光強度隨著時間的變化如圖9所示。(用天美(中國)科學儀器有限公司的熒光光譜儀F-7000進行測試得到)。
從圖9中可以看出,在紫外光線(UV)持續激發下,熒光強度幾乎沒有變化,表明本發明所述碳點可以穩定存在于UV下。
2碳點在自然光下的穩定性研究
取2mL的粘稠態碳點于石英比色皿中,將比色皿至于白光下,每隔一天就用光譜儀記錄下熒光強度。將光譜儀的雙狹縫調為5,電壓調為500V,激發波長與發射波長分別設為360nm和440nm,研究碳點熒光強度隨著時間的變化如圖10所示。(用天美(中國)科學儀器有限公司的熒光光譜儀F-7000進行測試得到)。
從圖10可以看出,在自然光激發下,熒光強度幾乎不變,表明本發明所述碳點可以穩定存在于自然光下。
上述實施例為本發明較佳的實施方式,但本發明的實施方式并不受上述實施例的限制,其他的任何未背離本發明的精神實質與原理下所作的改變、修飾、替代、組合、簡化,均應為等效的置換方式,都包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!