偶氮甲堿橋連噻吩化合物及其制備方法與流程
本發明涉及一種偶氮甲堿橋連噻吩化合物及其制備方法,適用于有機發光二極管、場效應管、柔性有源矩陣顯示、有機射頻電子商標、有機傳感器/存儲器、有機功能塑料、電子紙和太陽能電池等領域。
背景技術:
相比于無機半導體材料,有機半導體具有種類繁多、化學結構易于修飾的特點,有利于分子結構的設計與構造。有機半導體材料成膜性好,可用于制造大面積、柔性基材的光電器件。因而,有機半導體材料在低成本制造、有機傳感器、存儲器、集成電路等方面有著巨大的應用前景。
有機小分子半導體易于純化,減少雜質對器件性能的影響;同時,共軛小分子通常具有較好的共平面結構,有利于降低分子勢壘,獲得電荷載流子的高速遷移。低聚噻吩及其衍生物是研究報道較多的小分子半導體材料,噻吩環具有易于修飾和良好的載流子傳輸能力的特點[c.-a.di,y.q.liu,g.yu,d.b.zhu,acc.chem.res.2009,42,1573;d.demeter,j.roncali,s.jungsuttiwong,f.melchiorre,p.biagini,r.po,j.mater.chem.c,2015,3,7756.]。共軛單元橋連數個噻吩環是其中的一類,例如雙鍵、叁鍵、共軛基團等橋連聯噻吩衍生物等,表現出良好的性質[a.seidler,j.svoboda,v.dekoj,j.v.
偶氮甲堿(-n=ch-)是一種碳、氮原子雙鍵連接的共軛單元,偶氮甲堿單元與其碳類似物,即碳碳雙鍵具有等電子體的性質[c.wang,s.shieh,e.legoffandm.g.kanatzidis,macromolecules,1996,29,3147],且合成上易于實現,使得偶氮甲堿單元可以成為碳碳雙鍵的替代單元。
相對于碳碳雙鍵單元及其衍生物的研究,偶氮甲堿類化合物的研究滯后很多,其主要原因之一在于化學穩定的氨基取代噻吩衍生物前體數目很少,氨基取代噻吩中的氨基易于氧化,較難穩定存在[s.dufresneandw.g.skene,polym.mater.:sci.eng.,2005,92,16;w.g.skene,polym.prepr.(am.chem.soc.,div.polym.chem.),2004,45,252;c.-h.huang,n.d.mcclenaghan,a.kuhn,j.w.hofstraat,d.m.bassani,org.lett.,2005,7,3409.]。
技術實現要素:
本發明的目的是提供一種含偶氮甲堿橋連噻吩化合物及其制備方法。
本發明提供的偶氮甲堿橋連噻吩化合物(簡稱pntx=yr1r2r3r4),其結構通式如式i所示
所述式i中,所述r1均為碳原子總數為1-14的直鏈烷基或碳原子總數為10-24的支鏈烷基或為碳原子總數為3-12的直鏈烷基酯基;所述r2,r3,r4均為h原子或者親電子性基團,并且r2,r3,r4至少一個具有親電子性基團,所述親電子性基團含有氰基、鹵素或三氟甲基;所述的x為碳原子、y為氮原子,或者x為氮原子、y為碳原子;m為0-1的整數。
優選的,所述r1中,所述碳原子總數為1-14的直鏈烷基為甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、壬基、癸基、十一烷基、十二烷基、十三烷基、十四烷基;所述r1優選己基(pntx=yc6r2r3r4),癸基(pntx=yc10r2r3r4)。
優選的,所述r1中,所述碳原子總數為3-12的直鏈烷基酯基為羧酸甲酯、羧酸乙酯、羧酸丙酯、羧酸丁酯、羧酸戊酯、羧酸己酯、羧酸庚酯、羧酸辛酯、羧酸壬酯、羧酸癸酯;所述r1優選羧酸乙酯(pntx=yco2etr2r3r4)。
優選的,所述r2中,所述親電子性基團為鹵素、氰基、硝基、三氟甲基;所述r2優選氰基(pntx=yr1cnr3r4)。
優選的,所述r3中,所述為h原子或者鹵素、氰基、硝基、三氟甲基;所述的r3優選h原子(pntx=yr1r2hr4),溴原子(pntx=yr1r2brr4)。
優選的,所述r4中,所述為h原子或者鹵素、氰基、硝基、三氟甲基;所述的r4優選h原子(pntx=yr1r2r3h)
所述的m為0-1的整數,更優選m為0和1。
優選的,所述x、y中,所述的x為碳原子,同時y為氮原子;或者x為氮原子,同時y為碳原子。
本發明提供一種偶氮甲堿橋連噻吩化合物(式i)的制備方法,具體技術方案如下。
一種偶氮甲堿橋連噻吩化合物的制備方法,包括如下步驟:
所有制備的最終產物都是經由噻吩衍生物為原料,由氨基取代噻吩衍生物與醛基取代噻吩衍生物發生脫水縮合反應得到最終產物。
上述一種偶氮甲堿橋連噻吩化合物的制備方法的合成路線如下,可以制備不對稱結構的偶氮甲堿橋連噻吩化合物和對稱結構的偶氮甲堿橋連噻吩化合物。路線一中x為碳原子,y為氮原子。
路線二中x為氮原子,y為碳原子。
上述不對稱結構的偶氮甲堿橋連噻吩化合物的制備方法,合成方法如下:
在空氣或氮氣氣氛下,將等摩爾質量的氨基取代噻吩衍生物和醛基取代噻吩衍生物溶解于有機溶劑中,加入5-10mol%催化劑,室溫或者升溫攪拌,反應0.5-36小時,直到tlc確認原料反應完,真空條件下除去有機溶劑,得到粗產物,過柱或重結晶純化。
上述對稱結構的偶氮甲堿橋連噻吩化合物的制備方法,合成方法如下:
在空氣或氮氣氣氛下,將2摩爾當量醛基取代噻吩衍生物和1摩爾當量的氨基取代噻吩衍生物(路線一)或者2摩爾當量氨基取代噻吩衍生物和1摩爾當量的醛基取代噻吩衍生物(路線二)混合,溶于有機溶劑中,加入5-10mol%催化劑,室溫或者升溫攪拌,反應0.5-125小時,tlc確認原料反應完全,真空條件下除去有機溶劑,得到粗產物,過柱或重結晶純化。
上述不對稱結構和對稱結構的偶氮甲堿橋連噻吩化合物的制備方法中,所述有機溶劑,可優選醇溶劑(不局限于無水甲醇、無水乙醇、異丙醇、正丁醇)、苯、甲苯、無水甲苯、dmf、無水dmf、dmso、無水dmso、thf、無水thf等。
上述不對稱結構和對稱結構的偶氮甲堿橋連噻吩化合物的制備方法中,所述催化劑,可以不添加或者優選三氟乙酸、乙酸、硫酸、鹽酸、1,3-雙(二苯基膦)丙烷氯化鎳、四氯化鈦等。
上述不對稱結構和對稱結構的偶氮甲堿橋連噻吩化合物的制備方法中,所述溫度,可優選25-120℃。
上述偶氮甲堿橋連噻吩化合物在有機發光二極管、場效應管和太陽能電池中的應用。本發明的偶氮甲堿橋連噻吩化合物,偶氮甲堿與碳碳雙鍵碳類似物具有等電子性,柔性取代基的存在改善了化合物的溶解性,功能性官能團改善反應用前體的穩定性,并改變最終化合物的能級狀態。同時,碳氮雙鍵的存在為烷基鏈的排列提供了更大的空間,有利于烷基鏈的自組裝排列。因此可以作為載流子傳輸化合物廣泛地應用于電子器件中,如作為半導體材料,可作為有機發光二極管(oled)、場效應管(fet)和太陽能電池的有效元件。
本發明中,以上合成的化合物已經通過1hnmr、13cnmr、uv-vis、maldi-tof等手段進行了結構表征,確認了結構。
本發明提供了偶氮甲堿橋連噻吩化合物的制備方法,首次實現了這類化合物的制備;通過選擇不同氨基取代噻吩衍生物與醛基取代噻吩衍生物的脫水縮合制備化合物,方法簡單且產率高易于后續提純。本發明的化合物可作為載流子傳輸化合物廣泛地應用于電子器件中,如作為半導體材料,可作為有機發光二極管(oled)、場效應管(fet)和太陽能電池的有效元件。
下面通過附圖和具體實施方式對本發明做進一步說明,但并不意味著對本發明保護范圍的限制。
附圖說明
圖1-a至圖1-b為系列化合物(p3tc=nr1r2r3r4)的熱失重分析(tg)圖,圖1-a:p3tc=nc6cn;圖1-b:p3tc=nc10cn。
圖2-a至圖2-b為系列化合物(p3tc=nr1r2r3r4)的紫外可見吸收光譜(uv-vis)圖,圖2-a:p3tc=nc6cn;圖2-b:p3tc=nc10cn。
圖3-a至圖3-d為系列化合物(p3tn=cr1r2r3r4,p2tn=cr1r2r3r4)的熱失重分析(tg)圖,圖3-a:p3tn=cco2eth;圖3-b:p3tn=cco2etbr;3-c:p2tn=cco2eth;圖3-d:p2tn=cco2etbr。
圖4-a至圖4-d為系列化合物(p3tn=cr1r2r3r4,p2tn=cr1r2r3r4)的紫外可見吸收光譜(uv-vis)圖,圖4-a:p3tn=cco2eth;圖4-b:p3tn=cco2etbr;圖4-c:p2tn=cco2eth;圖4-d:p2tn=cco2etbr。
圖5為t-c6的核磁共振氫譜(1hnmr)圖。
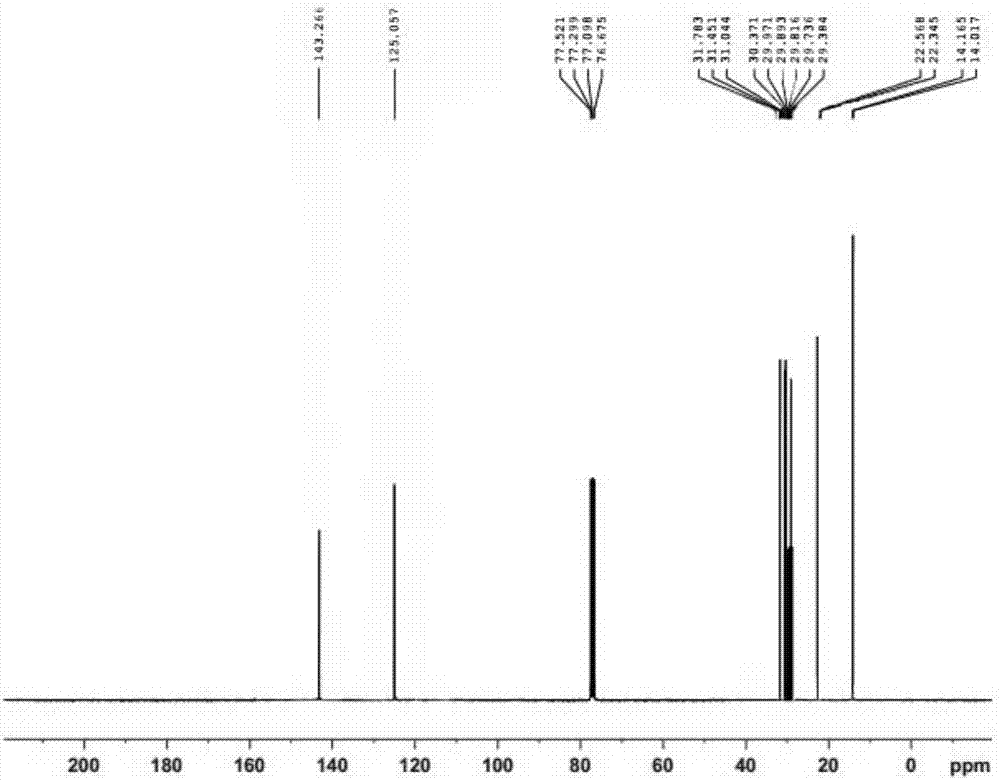
圖6為t-c6的核磁共振碳譜(13cnmr)圖。
圖7為t-c6的質譜(ms)圖。
圖8為t-c6-cho的核磁共振氫譜(1hnmr)圖。
圖9為t-c6-cho的核磁共振碳譜(13cnmr)圖。
圖10為t-c6-cho的質譜(ms)圖。
圖11為p3tc=nc6cn的核磁共振氫譜(1hnmr)圖。
圖12為p3tc=nc6cn的核磁共振碳譜(13cnmr)圖。
圖13為p3tc=nc6cn的質譜(ms)圖。
圖14為t-c10的核磁共振氫譜(1hnmr)圖。
圖15為t-c10的核磁共振碳譜(13cnmr)圖。
圖16為t-c10的質譜(ms)圖。
圖17為t-c10-cho的核磁共振氫譜(1hnmr)圖。
圖18為t-c10-cho的核磁共振碳譜(13cnmr)圖。
圖19為t-c10-cho的質譜(ms)圖。
圖20為p3tc=nc10cn的核磁共振氫譜(1hnmr)圖。
圖21為p3tc=nc10cn的核磁共振碳譜(13c。nmr)圖。
圖22為p3tc=nc10cn的質譜(ms)圖。
圖23為p3tn=cco2etbr的核磁共振氫譜(1hnmr)圖。
圖24為p3tn=cco2etbr的核磁共振碳譜(13cnmr)圖。
圖25為p3tn=cco2etbr的質譜(ms)圖。
圖26為p2tn=cco2etbr的核磁共振氫譜(1hnmr)圖。
圖27為p2tn=cco2etbr的核磁共振碳譜(13cnmr)圖。
圖28為p2tn=cco2etbr的質譜(ms)圖。
具體實施方式
下面結合具體實施例對本發明作進一步闡述,但本發明并不限于以下實施例。所述方法如無特別說明均為常規方法。所述反應物如無特別說明均能從公開商業途徑而得。
實施例1:制備2,2'-(((3,4-二己基噻吩-2,5-二基)雙(亞甲基)氨基)噻吩)二(噻吩-3-甲腈)(p3tc=nc6cn)。
(1)3,4-二己基噻吩的制備
在氮氣保護下,3,4-二溴噻吩(2.45g,10mmol)溶于無水四氫呋喃(75ml)中,加入催化劑1,3-雙(二苯基膦)丙烷氯化鎳(0.1g,0.2mmol)。置換氮氣,使用恒壓滴液漏斗滴加己基溴化鎂(34ml,34mmol),回流18小時。反應完畢后,使用砂芯漏斗墊硅藻土抽濾。所得有機相使用10%稀鹽酸洗,水洗,無水硫酸鈉干燥,過濾,減壓蒸去溶劑,通過柱層析提純得到3,4-二己基噻吩(無色油狀液體)1.27g,產率50%。
通過核磁分析,得到的是t-c6,具體數據為:1hnmr(300mhz,cdcl3):δ(ppm)=6.98(s,2h),2.72-2.55(m,4h),1.69(s,4h),1.39(s,12h),0.98(s,6h);13cnmr(75mhz,cdcl3):δ(ppm)=143.2,125.0,31.7,31.4,31.0,30.3,29.9,29.8,29.7,29.3,22.5,22.3,14.1,14.0。ms(ei-ms):m/z=252[m+h]+(calcd.forc16h28s:252.4)。圖5為t-c6的核磁共振氫譜(1hnmr)圖,圖6為t-c6的核磁共振碳譜(13cnmr)圖,圖7為t-c6的質譜(ms)圖。
(2)3,4-二己基噻吩-2,5-二甲醛的制備
在氮氣保護下,3,4-二己基噻吩(0.58g,2.31mmol)及四甲基乙二胺(1ml,6mmol)溶于無水正己烷(15ml)中,滴加正丁基鋰(3.75ml,6mmol),回流1.5小時。加入無水四氫呋喃(10ml),并降溫至―50℃。滴加無水dmf(1ml,13.5mmol),室溫攪拌2.5小時。反應完畢后,倒入水中,乙醚萃取,有機相無水硫酸鈉干燥,過濾,減壓蒸去溶劑,通過柱層析提純得到3,4-二己基噻吩-2,5-二甲醛(無色油狀液體)0.29g。產率42%。
通過核磁分析,得到的是t-c6-cho,具體數據為:1hnmr(300mhz,cdcl3):δ(ppm)=10.11(s,2h),2.92-2.87(t,4h),1.63-1.53(m,4h),1.43-1.32(m,12h),0.91-0.87(t,6h);13cnmr(75mhz,cdcl3):δ(ppm)=183.3,152.4,151.7,143.2,32.1,31.4,29.5,29.2,27.4,26.9,26.6,22.5,14.0。ms(ei-ms):m/z=308[m+h]+(calcd.forc18h28o2s:308.4)。圖8為t-c6-cho的核磁共振氫譜(1hnmr)圖,圖9為t-c6-cho的核磁共振碳譜(13cnmr)圖,圖10為t-c6-cho的質譜(ms)圖。
(3)2,2'-(((3,4-二己基噻吩-2,5-二基)雙(亞甲基)氨基)噻吩)二(噻吩-3-甲腈)的制備
將3,4-二己基噻吩-2,5-二甲醛(64.7mg,0.21mmol)及2-氨基噻吩-3-甲腈(132.68mg,1.07mmol)溶于正丁醇,120℃回流48小時。反應完畢后,減壓蒸去溶劑,通過柱層析提純得到產物2,2'-(((3,4-二己基噻吩-2,5-二基)雙(亞甲基)氨基)噻吩)二(噻吩-3-甲腈)(暗紅色固體)85.3mg,產率75%。
通過核磁分析,得到的是p3tc=nc6cn,具體數據為:1hnmr(300mhz,cdcl3):δ(ppm)=8.69(s,2h),7.15-7.10(m,4h),2.87-2.84(d,4h),1.57(s,4h),1.43-1.26(m,12h),0.89(s,6h);13cnmr(75mhz,cdcl3):δ(ppm)=162.9,152.3,150.1,140.8,128.2,121.9,114.5,104.7,31.7,31.5,29.3,27.2,22.5,14.0。ms(maldi-tof):m/z=521.1[m+h]+(calcd.forc18h32n4s3:520.7)。圖11為p3tc=nc6cn的核磁共振氫譜(1hnmr)圖,圖12為p3tc=nc6cn的核磁共振碳譜(13cnmr)圖,圖13為p3tc=nc6cn的質譜(ms)圖。
實施例2:制備2,2'-(((3,4-二癸基噻吩-2,5-二基)雙(亞甲基)氨基)噻吩)二(噻吩-3-甲腈)(p3tc=nc10cn)。
(1)3,4-二癸基噻吩的制備
在氮氣保護下,3,4-二溴噻吩(4.89g,20mmol)溶于無水四氫呋喃(150ml)中,加入催化劑1,3-雙(二苯基膦)丙烷氯化鎳(0.2g,0.4mmol)。置換氮氣,使用恒壓滴液漏斗滴加癸基溴化鎂(67ml,67mmol),回流18小時。反應完畢后,使用砂芯漏斗墊硅藻土抽濾。所得有機相使用10%稀鹽酸洗,水洗,無水硫酸鈉干燥,過濾,減壓蒸去溶劑,通過柱層析提純得到3,4-二癸基噻吩(無色油狀液體)4.23g,產率58%。
通過核磁分析,得到的是t-c10,具體數據為:1hnmr(300mhz,cdcl3):δ(ppm)=6.89(s,2h),2.53-2.48(t,4h),1.62(s,4h),1.28(s,28h),0.89(s,6h);13cnmr(75mhz,cdcl3):δ(ppm)=143.2,142.1,119.8,119.7,31.9,30.5,30.2,29.7,29.6,29.5,29.3,28.8,22.6,14.0。ms(ei-ms):m/z=364[m+h]+(calcd.forc24h44s:364.6)。圖14為t-c10的核磁共振氫譜(1hnmr)圖,圖15為t-c10的核磁共振碳譜(13cnmr)圖,圖16為t-c10的質譜(ms)圖。
(2)3,4-二癸基噻吩-2,5-二甲醛的制備
在氮氣保護下,3,4-二癸基噻吩(0.85g,2.31mmol)及四甲基乙二胺(1ml,6mmol)溶于無水正己烷(15ml)中,滴加正丁基鋰(3.75ml,6mmol),回流1.5小時。加入無水四氫呋喃(10ml),并降溫至―50℃。滴加無水dmf(1ml,13.5mmol),室溫攪拌2.5小時。反應完畢后,倒入水中,乙醚萃取,有機相無水硫酸鈉干燥,過濾,減壓蒸去溶劑,通過柱層析提純得到3,4-二癸基噻吩-2,5-二甲醛(無色油狀液體)0.39g。產率40%。
通過核磁分析,得到的是t-c10-cho,具體數據為:1hnmr(300mhz,cdcl3):δ(ppm)=10.12(s,2h),2.93-2.88(t,4h),1.64-1.56(m,4h),1.45-1.27(m,28h),0.90-0.86(t,6h);13cnmr(75mhz,cdcl3):δ(ppm)=183.2,151.6,143.2,32.1,31.8,29.6,29.5,29.4,29.2,26.6,22.6,14.0。ms(ei-ms):m/z=420[m+h]+(calcd.forc26h44o2s:420.7)。圖17為t-c10-cho的核磁共振氫譜(1hnmr)圖,圖18為t-c10-cho的核磁共振碳譜(13cnmr)圖,圖19為t-c10-cho的質譜(ms)圖。
(3)2,2'-(((3,4-二癸基噻吩-2,5-二基)雙(亞甲基)氨基)噻吩)二(噻吩-3-甲腈)的制備
將3,4-二癸基噻吩-2,5-二甲醛(88.2mg,0.21mmol)及2-氨基噻吩-3-甲腈(132.68mg,1.07mmol)溶于正丁醇,120℃回流48小時。反應完畢后,減壓蒸去溶劑,通過柱層析提純得到產物2,2'-(((3,4-二癸基噻吩-2,5-二基)雙(亞甲基)氨基)噻吩)二(噻吩-3-甲腈)(暗紅色固體)102mg,產率77%。
通過核磁分析,得到的是p3tc=nc10cn,具體數據為:1hnmr(300mhz,cdcl3):δ(ppm)=8.69(s,2h),7.14-7.11(m,4h),2.85(t,4h),1.60(s,4h),1.43-1.27(m,28h),0.88(s,6h);13cnmr(75mhz,cdcl3):δ(ppm)=162.9,152.6,152.3,151.7,150.1,140.9,140.8,128.2,122.0,121.9,121.5,114.5,104.7,31.8,31.7,29.6,29.5,29.3,28.9,27.2,26.9,25.2,22.6,14.1。ms(maldi-tof):m/z=633[m+h]+(calcd.forc26h48n4s3:632.3)。圖20為p3tc=nc10cn的核磁共振氫譜(1hnmr)圖,圖21為p3tc=nc10cn的核磁共振碳譜(13cnmr)圖,圖22為p3tc=nc10cn的質譜(ms)圖。
刪除該式子刪除該式子實施例3:制備2,5-雙(((5-溴噻吩-2-基)亞甲基)氨基)噻吩-3,4-二羧酸二乙酯(p3tn=cco2etbr)
(1)2,5-雙(((5-溴噻吩-2-基)亞甲基)氨基)噻吩-3,4-二羧酸二乙酯的制備
氮氣保護下,5-溴噻吩-2-甲醛(0.25mmol,47.75mg)溶于無水甲苯中,加入dabco(0.25mmol,29mg)(三乙烯二胺)后,冷卻至0℃,滴加四氯化鈦(1m甲苯溶液中)(0.25mmol),加入2,5-二氨基噻吩-3,4-二羧酸二乙酯(0.13mmol,33mg),回流24小時。反應完畢后,減壓蒸去溶劑,通過柱層析提純得到產物2,5-雙(((5-溴噻吩-2-基)亞甲基)氨基)噻吩-3,4-二羧酸二乙酯(紅色固體)。產率40%。
通過核磁分析,得到的是p3tn=cco2etbr,具體數據為:1hnmr(300mhz,cdcl3):δ(ppm)=8.31(s,2h),7.26-7.23(t,2h),7.10-7.08(d,2h),4.42-4.37(t,4h),1.43-1.38(t,6h);13cnmr(75mhz,cdcl3):δ(ppm)=163.0,150.9,148.6,143.5,133.5,131.2,127.6,121.1,61.4,14.3。ms(maldi-tof):m/z=604[m+h]+(calcd.forc20h16br2n2o4s3:604.3)。圖23為p3tn=cco2etbr的核磁共振氫譜(1hnmr)圖,圖24為p3tn=cco2etbr的核磁共振碳譜(13cnmr)圖,圖25為p3tn=cco2etbr的質譜(ms)圖。
實施例4:制備2-氨基-5-(((5-溴噻吩-2-基)亞甲基)氨基)噻吩-3,4-二羧酸二乙酯(p2tn=cco2etbr)
(1)2-氨基-5-(((5-溴噻吩-2-基)亞甲基)氨基)噻吩-3,4-二羧酸二乙酯的制備
將2,5-二氨基噻吩-3,4-二羧酸二乙酯(10mmol,2.58g)溶于乙醇中,加入5-溴噻吩-2-甲醛(15mmol,2.87g)攪拌。向瓶中加入催化劑三氟乙酸(1.67mmol,0.19g),回流反應16小時。反應完成后,減壓蒸去溶劑,通過柱層析提純得到產物2-氨基-5-(((5-溴噻吩-2-基)亞甲基)氨基)噻吩-3,4-二羧酸二乙酯(黃棕色晶體)。產率45%。
通過核磁分析,得到的是p2tn=cco2etbr,具體數據為:1hnmr(300mhz,cdcl3):δ(ppm)=7.93(s,h),7.11-7.02(m,2h),6.33(s,2h),4.44-4.37(m,2h),4.28-4.21(m,2h),1.47-1.42(t,3h),1.33-1.29(t,3h)。13cnmr(75mhz,cdcl3):δ(ppm)=165.1,164.3,159.5,144.6,144.1,133.3,130.8,130.7,129.9,118.3,103.1,61.5,60.3,14.4,14.1。ms(ei-ms):m/z=432[m]+(calcd.forc15h15brn2o4s2:431.3)。圖26為p2tn=cco2etbr的核磁共振氫譜(1hnmr)圖,圖27為p2tn=cco2etbr的核磁共振碳譜(13cnmr)圖,圖28為p2tn=cco2etbr的質譜(ms)圖。
本發明提供了偶氮甲堿橋連噻吩化合物的制備方法。本發明的制備方法成功的合成了對稱與不對稱偶氮甲堿橋連噻吩化合物。通過氨基取代噻吩衍生物與醛基取代噻吩衍生物發生脫水縮合反應得到產物,合成方法簡單。本發明的偶氮甲堿橋連噻吩化合物與碳碳雙鍵類似物具有等電子性。柔性取代基的存在改善了化合物的溶解性,功能性官能團改善反應用前體的穩定性,并改變最終化合物的能級狀態。同時,碳氮雙鍵橋的存在為烷基鏈的排列提供了更大的空間,有利于烷基鏈的自組裝排列。因此可以作為載流子傳輸化合物廣泛地應用于電子器件中,如作為半導體材料,可作為有機發光二極管(oled)、場效應管(fet)和太陽能電池的有效元件。
最后說明的是,以上實施例僅用以說明本發明的技術方案而非限制,盡管參照最佳實施例對本發明進行了詳細說明,本領域的普通技術人員應當理解,可以對本發明的技術方案進行修改或等同替換,而不脫離本發明技術方案的宗旨和范圍,其均應涵蓋在本發明的權利要求范圍當中。
- 還沒有人留言評論。精彩留言會獲得點贊!