預靶向試劑盒、方法和其中使用的試劑與流程
本發明涉及用于靶向的醫學成像和/或治療的預靶向方法(pretargetingmethod),其中使用彼此顯示出生物正交(bio-orthogonal)反應性的非生物(abiotic)反應性化學基團。本發明還涉及含有至少一種預靶向探針(pre-targetingprobe)和至少一種效應物探針(effectorprobe)的預靶向試劑盒(kit),其中所述預靶向探針包含初級靶向部分(primarytargetingmoiety)和第一生物正交反應性基團,和其中所述效應物探針包含效應物部分,例如標記物或藥學活性化合物,和第二生物正交反應性基團。本發明還涉及在上述方法和試劑盒中使用的預靶向試劑。本發明特別地涉及核成像和放射療法。
背景技術:
在醫學診斷和治療的許多領域中,希望選擇性地將試劑,例如治療劑(藥物)或診斷(例如成像)劑遞送到受試者(subject)例如病人體內的特定位點或有限區域。器官或組織的活性靶向通過所希望的活性部分(例如對比增強劑或細胞毒素化合物)對靶向結構的直接或間接結合實現,其與細胞表面結合或促進在感興趣的靶位點處或附近的細胞攝取。用于靶向此類試劑的靶向部分典型地是對細胞表面靶(例如膜受體)、結構蛋白(例如淀粉樣斑)或細胞內靶(例如RNA、DNA、酶、細胞信號通路)具有親和性的結構。這些部分可以是抗體(片段)、蛋白質、適體(aptamers)、寡肽、寡核苷酸、寡糖以及肽、擬肽(peptoids)和已知在特定疾病或障礙處積累的有機藥物化合物。作為選擇,對比/治療劑可以靶向代謝途徑,其在疾病(例如感染或癌癥)期間表達升高,例如DNA、蛋白質和膜合成和碳水化合物攝取。在患病組織中,上述標志物可以將患病細胞從健康組織區分并提供早期檢測、特異性診斷和(靶向的)治療的獨特可能性。通常對于成功的分子成像/治療劑和特別對于核成像/治療劑一個重要標準是它們呈現高靶攝取,同時顯示出從非靶組織和從血液的快速清除(通過腎臟和/或肝膽體系)。但是,這經常存在問題:例如,人中的成像研究已經顯示,在腫瘤位置處經放射標記的抗體在24小時內可達到最大濃度,但需要另外幾天在循環中的所述經標記的抗體的濃度才下降到低至足以發生成功成像的水平。在靶組織中的緩慢或不充分積累以及從非靶區域緩慢清除所帶來的這些問題(特別是對于核成像和治療)已經導致預靶向方法的應用。預靶向是指在靶向方法中的步驟,其中向初級靶(例如細胞表面)提供有預靶向探針。后者包括次級靶,其最后將通過裝備有次級靶向部分的進一步的探針(所述效應物探針)進行靶向。這樣,在預靶向中,將預靶向探針結合到初級靶。所述預靶向探針還攜帶次級靶,其促進對診斷(成像)和/或治療劑,所述效應物探針的特異性結合。在形成所述預靶向探針的結構已經定位在靶位點處之后(花費時間例如24小時),如果自然清除不充分,則可以使用清除劑從血液除去過量的部分。在第二培育步驟(優選采取較短的時間,例如1-6小時)中,所述效應物探針通過它的次級靶向部分(secondarytargetingmoiety)結合到所述(預)結合的預靶向探針。所述次級靶(存在于所述預靶向探針上)和所述次級靶向部分(存在于所述效應物探針上)應該以高特異性和高親和性快速結合,并且在體內應該是穩定的。對于成像,預靶向的一般概念在圖1中繪出。在這里,所述效應物探針是包含用于成像模式的可檢測標記物的成像探針。所述效應物探針通過其次級靶向基團結合到所述(預)結合的預靶向探針。次級靶/次級靶向部分對的一般例子是生物素/抗生蛋白鏈菌素或抗體/抗原體系。為了有效,所述效應物探針必需快速從體內排出(例如通過腎臟)以提供所希望的具有相對低的非靶積累的高腫瘤積累。因此,這些探針通常很小。在核成像和放射療法中,預靶向的概念具有進一步的益處,因為花費時間的預靶向步驟可以不必使用放射性核素實施,而使用放射性核素的次級靶向步驟能夠更快地實施。后者使得能夠使用較短壽命的放射性核素,具有將對病人的放射劑量最小化的優點,例如使用PET試劑代替SPECT試劑。結合多齒配體體系(抗生蛋白鏈菌素、樹枝狀聚合物(dendrimers))在MRI中使用預靶向方法能夠在靶位點處提供信號放大。此外,通常該方法有利于通用對比劑的使用。通常在生物學中(如抗體-抗原)和特別在預靶向中(生物素-抗生蛋白鏈菌素、抗體/半抗原、反義寡核苷酸)實施高度選擇性相互作用的實體非常大。因此,使用肽和小有機部分作為初級靶向基團的預靶向,以及代謝成像和細胞內靶成像,由于次級靶的尺寸使得小初級基團的使用毫無意義而仍然無法實現。而且,現有的預靶向體系被與它們的生物特性相關的因素所限制。生物素是內源分子,并且它的綴合物(conjugates)能夠通過血清酶生物素酶斷開。當使用反義預靶向時,所述寡核苷酸會受到RNAse和DNAse的攻擊。蛋白質和肽也經歷自然分解途徑。這些相互作用可被它們的非共價和動態特性以及有限的靶上駐留時間進一步削弱。而且,內源性生物素與生物素綴合物競爭抗生蛋白鏈菌素結合。最后,抗生蛋白鏈菌素是高度免疫原性的。近來的一個發展是避免與僅基于天然/生物靶向結構(即生物素/抗生蛋白鏈菌素、抗體/半抗原、反義寡核苷酸)的預靶向相關的缺點。這方面的一篇文獻是WO2010/051530,其中基于某些二烯例如四嗪類和親雙烯體例如反式-環辛烯醇(trans-cyclooctenol,TCO)之間的反應性對預靶向進行了討論。這方面的一篇進一步的文獻是Li等,ChemicalCommunications,2010,46(42),p.8043-8045,其描述了基于3,-二芳基-s-四嗪和18F-標記的反式-環辛烯之間的Diels-Alder反應的用于生物結合的放射標記方法。Rossin等,Angew.Chem.Int.,Ed2010,49,p.3375-3378涉及通過使用逆電子需求的Diels-Alder反應(inverse-electron-demandDiels-Alderreaction)的腫瘤預靶向。Blackman等,J.Am.Chem.Soc.,2008,130,p.13518-13519描述了基于逆電子需求的Diels-Alder反應性的快速生物結合。Royzen等,J.Am.Chem.Soc.,2008,130,p.3760-3761涉及通過金屬絡合驅動的官能化的反式-環辛烯的光化學合成。盡管在這些體系的基礎上能夠獲得相對快速的反應,但這還遠不如上述生物素-抗生蛋白鏈菌素體系的反應性。因此,避免后者的缺點,卻犧牲了所述反應的主要要求,即速度。因此,希望提供一種體系,其不以如上討論的生物分子為基礎,并且還具有理想的快速反應速率。
技術實現要素:
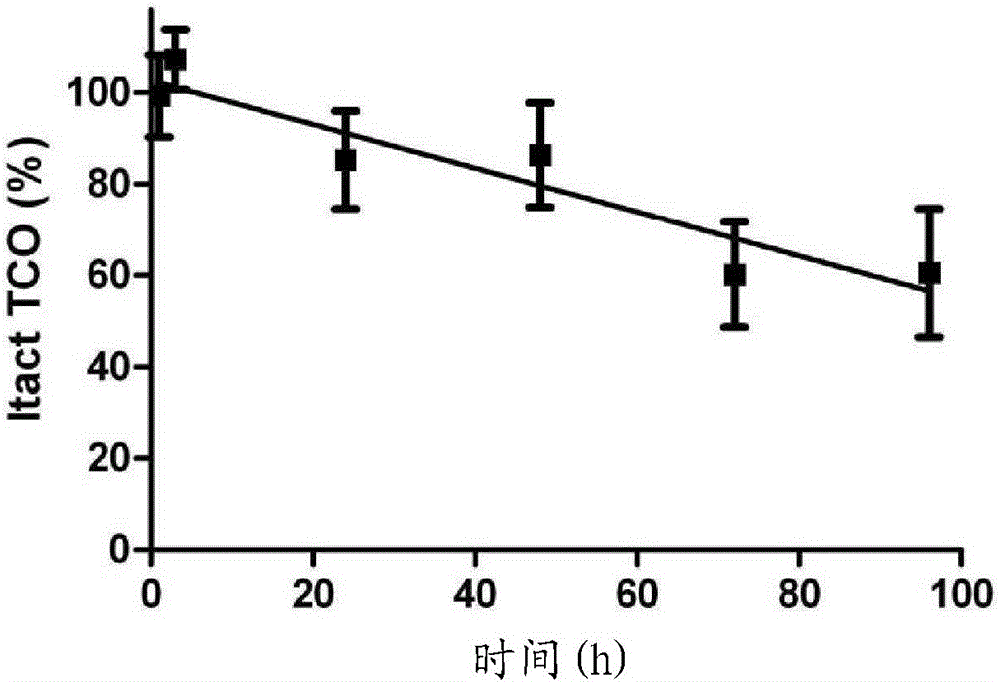
為了更好地滿足上述愿望,本發明一方面提供了用于靶向的醫學成像和/或治療的試劑盒(kit),其包含至少一種預靶向探針和至少一種效應物探針,其中所述預靶向探針包含初級靶向部分和第一生物正交反應性基團,和其中所述效應物探針包含效應物部分(例如標記物或藥學活性化合物)和第二生物正交反應性基團,其中所述第一和第二生物正交反應性基團的任一個是親雙烯體(dienophile),而所述第一和第二生物正交反應性基團中的另一個是二烯,其中所述親雙烯體是滿足式(1)的8元環親雙烯體:其中R的位置是平伏的(equatorial)和Ra的位置是直立的(axial),其中X、Y、R、和Ra的每一個獨立地代表H,或者最多六次(inatmostsixinstances)代表選自由以下組成的組的取代基:烷基、芳基、O-芳基、O-烷基、S-芳基、S-烷基、S(O)-芳基、S(O)-烷基、S(O)2-芳基、S(O)2-烷基、Si-芳基、Si-烷基、Si-O-烷基、OCO-烷基、OCO-芳基、SCO-烷基、SCO-芳基、OCS-烷基、OCS-芳基、SCS-烷基、SCS-芳基、F、Cl、Br、I、N3、SO2H、SO3H、SO4H、PO4H、OH、SH、NO2、NO、CN、OCN、SCN、NCO、NCS、CF3、NR’R”其中R’和R’’各自獨立地是H、烷基或芳基、C(=O)O-烷基、C(=O)O-芳基、C(=S)O-烷基、C(=S)O-芳基、C(=O)S-烷基、C(=O)S-芳基、C(=S)S-烷基、C(=S)S-芳基、C(=O)NR’R’’其中R’和R’’各自獨立地是H、芳基或烷基、NR’CO-烷基其中R’是H、烷基或芳基、NR’CO-芳基其中R’是H、烷基或芳基、NR’C(=O)O-烷基其中R’是H、烷基或芳基、NR’(C=O)O-芳基其中R’是H、烷基或芳基、OCONR’-烷基其中R’是H、烷基或芳基、OCONR’-芳基其中R’是H、烷基或芳基、NR’CONR’’-烷基其中R’和R’’各自獨立地是H、烷基或芳基、NR’CONR’’-芳基其中R’和R’’各自獨立地是H、烷基或芳基、NR’CSNR’’-烷基其中R’和R’’各自獨立地是H、烷基或芳基和NR’CSNR’’-芳基其中R’和R’’各自獨立地是H、烷基或芳基、CR’NR’’其中R’和R’’各自獨立地是H、烷基或芳基;其中Ra中的一個包含在任選地通過間隔體至所述預靶向探針或效應物探針的連接體部分(LinkerMoiety)中;其中兩個R或Ra部分可以一起形成環;和其中至少一個且最多四個Ra不是氫。另一方面,本發明提供了預靶向方法,以及其中使用的預靶向試劑,和其中使用該試劑盒的靶向的醫學成像或療法。再一方面,本發明是滿足上述式(1)的化合物,其用于動物或人類中的預靶向方法中。又一方面,本發明在于具有一個或多個直立取代基(axialsubstituents)的反式-環辛烯在基于逆Diels-Alder反應的預靶向方法中作為親雙烯體反應物的用途。附圖說明圖1描繪了如上所述的預靶向概念的一般方案。圖2提供了用于[4+2]Diels-Alder反應的反應方案;在(3,6)-二-(2-吡啶基)-s-四嗪和E-環辛烯之間,接著是其中形成產物和分子氮的逆Diels-Alder反應。因為所述反式-環辛烯衍生物不像經典Diels-Alder反應中那樣包含吸電子基團,所以這種類型的Diels-Alder反應與經典的Diels-Alder反應不同,并且通常稱為“逆電子需求Diels-Alder反應”。在下文中,兩個反應步驟序列即最初的Diels-Alder環加成反應(典型地是逆電子需求Diels-Alder環加成)和后續的逆Diels-Alder反應將簡稱為“逆Diels-Alder反應”或“逆DA”。圖3(a和b)描繪了利用逆Diels-Alder化學用于預靶向的一般方案。圖4提供了利用涉及TCO-改性的mAb(B)和放射標記的四嗪(A)的逆-DA的腫瘤預靶向方案。圖5到圖10示出了實施例中涉及的化合物的合成方案。圖11示出了環辛烯醇的以下討論的E-次要和E-主要異構體,以及Z異構體的對比,示出了立體化學。圖12示出了四嗪-DOTA探針28的結構。圖13描繪了三種不同的環辛烯親雙烯體在小鼠中的體內穩定性。圖14示出了活小鼠的SPECT/CT投影。圖15描繪了四嗪探針28的替代合成過程,和相應的Gd-絡合物,28-GdIII的合成。圖16示出了四嗪模型探針35、38、40和42的合成路線。圖17示出了新型TCOs的合成路線。圖18提供了在PBS中在低濃度下TCO(20a或20b)和載體加入的(carrieradded)177Lu-四嗪28(1eq.)之間的歸一化反應收率。圖19提供了對血液清除進行校正的CC49-結合的TCO20b的體內穩定性。所述數據點代表平均值和所述誤差棒代表一個標準偏差(n=3)。圖20示出了CC49-TCO(20b)結構的血液曲線。數據點代表平均值和誤差棒表示一個標準偏差(n=3)。圖21提供了無腫瘤小鼠中CC49-TCO(20b)結構的生物分布。柱代表平均值和誤差棒代表一個標準偏差(n=3)。圖22示出了125I-CC49和125I-CC49-TCO44b(7.5)在無腫瘤小鼠(n=3)中的血液動力學。數據以具有一個標準偏差(誤差棒)的百分比注射劑量/克(%ID/g)給出。圖23提供了mAb注射4天后125I-CC49(空心柱)和125I-CC49-TCO44b(7.5)(實心柱)在無腫瘤小鼠中(n=3)中的生物分布。所述柱代表具有一個標準偏差(誤差棒)的百分比注射劑量/克(%ID/g)。圖24示出了結合到CC49的TCO44b的體內穩定性。數據點是具有一個標準偏差(誤差棒)的三次測量的平均,擬合為二階多項式(PrismGraphPadv.5.01)。具體實施方式下文中以E-環辛烯表示本發明中使用的張力環辛烯(strainedcyclooctene)親雙烯體。根據常規命名法,將理解作為取代X或Y的結果,取決于取代基的位置和分子量,相同的環辛烯異構體形式上可以變成表示為Z-異構體。在本發明中,本發明的任何取代變體,無論是否形式上是“E”或“Z”,或“順式”或“反式”異構體,都將被認為是未取代的反式-環辛烯,或未取代的E-環辛烯的衍生物。術語“反式-環辛烯”(TCO)以及E-環辛烯可以互換使用并且對于根據本發明的所有親雙烯體,以及對于取代基形式上將需要相反的命名的情況,都適用。即本發明涉及其中如下編號的碳原子1和6位于E(entgegen)或反式位置的環辛烯。在一般意義上,本發明基于這樣一個認識:在使用衍生的反式-環辛烯,例如以反式-環辛烯醇的形式,作為親雙烯體的體系中,選擇其中用于衍生成連接體結構的羥基處于直立位置的異構體。E-環辛烯醇,其處于所謂的冠式構象(crownconformation),具有兩種異構體,在平伏位置(equatorialposition)具有OH的一種是主要異構體和在直立位置具有OH的一種是次要異構體。根據本發明選擇所述后一種異構體,并且在下文以“E-次要”表示。不希望被理論所限,本發明人相信,基于這一發現,TCO上一個或多個直立取代基的存在為解決基于所述逆Diels-Alder反應的預靶向中對于更高反應性的需求提供了答案。廣義上,本發明因此擴展到超出關于如上提供的式(1)給出的取代基定義。存在一個或多個直立取代基的事實據信導致較高的HOMO能量。HOMO,如本領域技術人員所知,代表最高占據分子軌道。根據本發明,使用MOPAC模擬來確定所述TCO的HOMO能量。MOPAC(MolecularOrbitalPackage,分子軌道包)是計算化學領域中公知的軟件。所述MOPAC軟件包包括幾種眾所周知的半經驗分子軌道方法,包括AM1和PM3。術語AM1和PM3是指不同的哈密頓函數(Hamiltonians),即AustinModel1和參數化的模型3哈密頓函數(對于AM1,還請見M.J.S.Dewar等,J.Am.Chem.Soc.,107,3902(1985);對于PM3,還請見J.J.P.Stewart,.J.Comput.Chem.,10,209(1989)和J.Comput.Chem.,10,221(1989))。在本發明中,使用包含以下的方法:在計算機環境中,提供環辛烯的衍生物的分子結構,通過確定最低形成能量優化各分子結構,確定所述AM1和PM3哈密頓函數并從而確定最高占據分子軌道(HOMO)。表6中的MOPAC數據顯示直立取代基的存在服務于在所述反式-環辛烯環中提供增加的HOMO能量的目的。將就特定的實施方案并參考某些附圖進一步描述本發明,但本發明并不受這些所限制而只由權利要求限制。權利要求中的任何附圖標記都不應解釋為限制范圍。所描述的附圖僅僅是示意性的而非限定性的。在所述附圖中,出于說明目的一些要素的尺寸可能被夸大并且不按比例畫出。在本說明書和權利要求中當使用術語“comprising”時,其并不排除其它要素或步驟。當涉及單數名詞使用不定冠詞或定冠詞例如“a”或“an”、“the”時,除非另外特別說明否則其包括多個(種)所述名詞。此外,還要注意說明書和權利要求中使用的術語“comprising”不應被解釋為限于隨后列舉的設備;其不排除其它要素或步驟。因此,表述“包含設備A和B的裝置”的范圍不應被限定為僅由組件A和B組成的裝置。其表示,就本發明而言,所述裝置的僅有相關組件是A和B。在幾個化學式中涉及“烷基”和“芳基”。在這方面,“烷基”各自獨立地表示不超過十個碳原子的脂肪族直鏈、支鏈或環狀烷基基團,其可包括1-3個雜原子例如O、N或S,優選具有1-6個碳原子,和“芳基”各自獨立地表示不超過十個碳原子的芳基或雜芳基,其可包括1-3個雜原子例如N或S。在幾個化學式中,關于字母例如“A”、“B”、“X”、“Y”和各種編號的“R”基團表示了基團或取代基。這些字母的定義將參考各式進行解讀,即在不同的式中,除非另外說明,否則這些字母可各自獨立地具有不同的意義。在本發明的進一步優選的實施方案中,在以下幾個化學式中涉及“烷基”和“芳基”。在這方面,“烷基”各自獨立地表示不超過10個碳原子的脂肪族的、直鏈的、支鏈的、飽和的、不飽和的和/或環狀的烴基,其可包括1-10個雜原子例如O、N、或S,和“芳基”各自獨立地表示不超過20個碳原子的芳基或雜環芳基,其可以是被取代的,并且其可以包括1-10個雜原子例如O、N、P或S。“芳基”還包括“烷芳基”或“芳烷基”(簡單的例子:芐基)。“烷基”、“芳基”、“烷芳基”和“芳烷基”包含的碳原子數可以通過此類術語前面的稱號表示(即C1-C10烷基指所述烷基可包含1到10個碳原子)。本發明的某些化合物具有手性中心和/或互變異構體,并且所有的對映異構體、非對映異構體(diasteriomers)和互變異構體,以及它們的混合物都在本發明的范圍之內。逆Diels-Alder反應逆Diels-Alder耦合化學通常包括耦合以形成不穩定中間體的反應物對,所述中間體通過逆Diels-Alder反應消除作為唯一副產物的小分子(取決于起始化合物其可以是例如N2、CO2、RCN)以形成穩定產物。所述成對反應物包括作為一種反應物(即一種生物正交反應性基團)的適合的二烯,例如四嗪衍生物如缺電子四嗪,和作為另一種反應物(即另一種生物正交反應性基團)的根據式(1)的張力(strained)環辛烯。例如缺電子(取代的)四嗪與本發明的張力E-環辛烯的異常快速的反應導致了連接反應中間體(ligationintermediate),其在[4+2]逆Diels-Alder環加成中通過消除作為唯一副產物的N2重排為穩定的二氫噠嗪。這在圖2中示出。這兩種反應性物類是非生物的,因此不經歷快速的體內代謝。它們是生物正交的,例如它們在生理介質中彼此選擇性地反應。關于這點的一個優點在于所述二烯和所述環辛烯都基本上對細胞內或細胞表面上的生物分子和所有其它區域如血清等是非反應性的。因此,本發明的化合物和方法能夠在活細胞、組織或生物體(organism)中使用。而且,所述反應性基團相對較小并且能夠引入生物試樣或活生物體中而不顯著改變生物學尺寸。利用所述[4+2]逆Diels-Alder反應,可以利用小的反應參與者例如四嗪或環辛烯將大尺寸的初級靶向部分例如抗體與標記物或其它分子結合。甚至更有利地,利用(匹配的)相對小的反應參與者例如四嗪和環辛烯,可以使相對小的初級靶向部分例如肽與標記物或其它分子結合。所述預靶向探針和效應物探針的尺寸和性質不受所述次級靶和次級靶向部分的大影響,使得(預)靶向方案能夠用于小的靶向部分。因為這樣,能夠靶向其它組織,即所述探針的目的地不限于脈管系統(vascularsystem)和胞間隙(目前使用抗體-抗生蛋白鏈菌素的靶向通常是限于脈管系統和胞間隙)。關于逆電子需求Diels-Alder反應和反應性物類對的行為的文獻包括:Thalhammer,F;Wallfahrer,U;Sauer,J,TetrahedronLetters,1990,31(47),6851-6854;Wijnen,JW;Zavarise,S;Engberts,JBFN,JournalOfOrganicChemistry,1996,61,2001-2005;Blackman,ML;Royzen,M;Fox,JM,JournalOfTheAmericanChemicalSociety,2008,130(41),13518-19),R.Rossin,P.RenartVerkerk,SandraM.vandenBosch,R.C.M.Vulders,l.Verel,J.Lub,M.S.Robillard,AngewChemIntEd2010,49,3375,N.K.Devaraj,R.Upadhyay,J.B.Haun,S.A.Hilderbrand,R.Weissleder,AngewChemIntEd2009,48,7013和Devaraj等,Angew.Chem.Int.Ed.,2009,48,1-5。應該理解,廣義上,根據本發明上述耦合化學基本上可以應用于預靶向中能夠使用的任何分子、基團或部分對。即一個這樣的對將包含初級靶向部分,其能夠結合到初級靶,和進一步包含至少一個次級靶。另一個將是適合用于結合到所述次級靶的次級靶向部分,和進一步包含適合發揮治療作用(典型地是藥學活性化合物),或適合通過成像技術定位(即標記物)或兩者的部分。因此,根據本發明,所述預靶向探針和所述效應物探針中的任一被上面限定的直立取代的環辛烯官能化,而另一個被四嗪或其它適合的二烯官能化。這在圖3中示出。上部的方案(圖3a)示出了預靶向探針,含有通過連接體部分(任選包含柔性間隔體)與作為初級靶向部分的抗體連接的二吡啶基四嗪,和效應物探針,含有通過連接體(柔性間隔體)與可檢測標記物連接的環辛烯(作為次級靶向部分)。下面的方案(圖3b)示出了正好相反的方案,即包含環辛烯的預靶向探針和包含四嗪的效應物探針。盡管所述圖中沒有清楚示出立體化學,應該理解,在本發明中所述環辛烯是依照如上定義的式(1)的直立取代的環辛烯。親雙烯體本發明的一個基本成就在于選擇親雙烯體,即根據如上定義的式(1)的直立取代的TCO,其使得對于所述生物正交耦合反應能夠獲得高達10倍或更多的增加的反應速率。或者換一種說法,反應時間僅是原來需要的時間的10%。或者再換一種說法,一種反應物的濃度可以低10倍。所述親雙烯體,廣義上,是具有至少一個直立取代基的反式-環辛烯,即其中至少一個飽和碳原子的至少一個直立位置不是氫。如以上解釋的那樣,至少一個且最多四個Ra不是氫,意味著這種Ra是取代基或是連接體結構的一部分。優選地,非氫Ra的數量是1或2。更優選地,所述至少一個和最多四個所述非氫Ra位于選自由R2a、R3a、R4a和R5a組成的組的位置。仍更優選地,R2a、R3a、R4a和R5a中的一個或兩個不是氫。最優選地,取代基或連接體結構作為R3a和R4a中的一個或這兩者存在。優選地,所述取代基選自以上關于如上定義的式(1)定義的基團。更優選地,上述Ra是烷基或O-烷基,更優選地是甲基或O-叔丁基。應該注意的是,Ra的選擇和優選選項與另一碳原子上或相同碳原子上在平伏位置是否存在任何取代基(即如上定義的式(1)中的R基團)無關。優選地,除了一個或兩個直立取代基以外,還存在一個或兩個平伏取代基,所述取代基優選包括是連接體結構的一部分的R或Ra。但是,在另一優選項中,考慮到在合成工作和反應性之間達成平衡,優選一個或兩個Ra不是氫,而所有其它R和Ra都是氫。在一個進一步的優選項中,X和/或Y是O-烷基或烷基,更優選地是甲基。二烯本領域技術人員知道在所述逆Diels-Alder反應中具有反應性的大量二烯。優選的二烯參考式(2)-(5)在下文給出。其中R1選自由以下組成的組:H、烷基、芳基、CF3、CF2-R’、OR’、SR’、C(=O)R’、C(=S)R’、C(=O)O-R’、C(=O)S-R’、C(=S)O-R’、C(=S)S-R’、C(=O)NR’R’’、C(=S)NR’R’’、NR’R’’、NR’C(=O)R’’、NR’C(=S)R’’、NR’C(=O)OR’’、NR’C(=S)OR’’、NR’C(=O)SR’’、NR’C(=S)SR’’、NR’C(=O)NR’’R’’’、NR’C(=S)N’R’’R’’’其中R’、R’’和R’’各自獨立地是H、芳基或烷基;A和B各自獨立地選自由烷基取代的碳、芳基取代的碳、氮、N+O-、N+R其中R為烷基組成的組,條件是A和B不都是碳;X選自由O、N-烷基和C=O組成的組,和Y是CR其中R選自由以下組成的組:H、烷基、芳基、C(=O)OR’、C(=O)SR’、C(=S)OR’、C(=S)SR’、C(=O)NR’R’’其中R’和R’’各自獨立地是H、芳基或烷基;對于環辛烯特別適合作為反應參與者的二烯是:其中R1和R2各自獨立地選自由以下組成的組:H、烷基、芳基、CF3、CF2-R’、NO2、OR’、SR’、C(=O)R’、C(=S)R’、OC(=O)R’’’、SC(=O)R’’’、OC(=S)R’’’、SC(=S)R’’’、S(=O)R’、S(=O)2R’’’、S(=O)2NR’R’’、C(=O)O-R’、C(=O)S-R’、C(=S)O-R’、C(=S)S-R’、C(=O)NR’R’’、C(=S)NR’R’’、NR’R’’、NR’C(=O)R’’、NR’C(=S)R’’、NR’C(=O)OR’’、NR’C(=S)OR’’、NR’C(=O)SR’’、NR’C(=S)SR’’、OC(=O)NR’R’’、SC(=O)NR’R’’、OC(=S)NR’R’’、SC(=S)NR’R’’、NR’C(=O)NR’R’’、NR’C(=S)N’R’R’’,其中R’和R’’各自獨立地是H、芳基或烷基,和R’’’獨立地是芳基或烷基;A選自由N-烷基、N-芳基、C=O和CN-烷基組成的組;B是O或S;X選自由以下組成的組:N、CH、C-烷基、C-芳基、CC(=O)R’、CC(=S)R’、CS(=O)R’、CS(=O)2R’’’、CC(=O)O-R’、CC(=O)S-R’、CC(=S)O-R’、CC(=S)S-R’、CC(=O)NR’R’’、CC(=S)NR’R’’,R’和R’’各自獨立地是H、芳基或烷基和R’’’獨立地是芳基或烷基;Y選自由CH、C-烷基、C-芳基、N和N+O-組成的組;對于環辛烯另一種特別適合作為反應參與者的二烯是:其中R1和R2各自獨立地選自由以下組成的組:H、烷基、芳基、CF3、CF2-R’、NO2、OR’、SR’、C(=O)R’、C(=S)R’、OC(=O)R’’’、SC(=O)R’’’、OC(=S)R’’’、SC(=S)R’’’、S(=O)R’、S(=O)2R’’’、S(=O)2NR’R’’、C(=O)O-R’、C(=O)S-R’、C(=S)O-R’、C(=S)S-R’、C(=O)NR’R’’、C(=S)NR’R’’、NR’R’’、NR’C(=O)R’’、NR’C(=S)R’’、NR’C(=O)OR’’、NR’C(=S)OR’’、NR’C(=O)SR’’、NR’C(=S)SR’’、OC(=O)NR’R’’、SC(=O)NR’R’’、OC(=S)NR’R’’、SC(=S)NR’R’’、NR’C(=O)NR’R’’、NR’C(=S)N’R’R’’,其中R’和R’’各自獨立地是H、芳基或烷基,和R’’’獨立地是芳基或烷基;A選自由N、C-烷基、C-芳基和N+O-組成的組;B是N;X選自由以下組成的組:N、CH、C-烷基、C-芳基、CC(=O)R’、CC(=S)R’、CS(=O)R’、CS(=O)2R’’’、CC(=O)O-R’、CC(=O)S-R’、CC(=S)O-R’、CC(=S)S-R’、CC(=O)NR’R’’、CC(=S)NR’R’’,其中R’和R’’各自獨立地是H、芳基或烷基和R’’’獨立地是芳基或烷基;Y選自由CH、C-烷基、C-芳基、N和N+O-組成的組;特別有用的四嗪衍生物是缺電子四嗪類,即被通常不被認為供電子的基團或部分取代的四嗪,優選帶有吸電子取代基。這些缺電子四嗪類通常符合以下結構式:在此,R1和R2各自獨立地代表選自由以下組成的組的取代基:H、2-吡啶基、3-吡啶基、4-吡啶基、2,6-嘧啶基、2,5-嘧啶基、3,5-嘧啶基、2,4-嘧啶基、或苯基,任選地被一個或多個吸電子基團例如NO2、F、Cl、CF3、CN、COOH、COOR、CONH2、CONHR、CONR2、CHO、COR、SO2R、SO2OR、NO、Ar取代,其中R是C1-C6烷基和Ar代表芳基,特別是苯基、吡啶基或萘基。在根據式(2)-(5)每一個的化合物中,所述R1和R2基團(包括在X或Y上的那些)可以進一步提供有如下所述的適合的連接體或間隔體部分。類似地,并且與其獨立地,如上定義的式(1)的親雙烯體也可以進一步提供有如下所述的適合的連接體或間隔體部分。根據一個實施方案,本發明被用于靶向的成像。根據該實施方案,特定的初級靶的成像通過所述預靶向探針的初級靶向部分的特異性結合以及利用所述效應物探針中包含的可檢測標記物檢測這種結合實現。初級靶本發明中使用的“初級靶(primarytarget)”涉及在診斷和/或成像方法中待被檢測,和/或待被通過藥學活性化合物或其它治療形式調制、結合或尋址的靶。所述初級靶可以選自人類或動物體內或病原體或寄生物上的任何適合的靶,例如包含以下的組:細胞例如細胞膜和細胞壁、受體例如細胞膜受體、細胞內結構例如高爾基體或線粒體、酶、受體、DNA、RNA、病毒或病毒顆粒、抗體、蛋白質、碳水化合物、單糖、多糖、細胞活素、荷爾蒙、類固醇、生長抑素受體、單胺氧化酶、毒蕈堿性受體、心肌交感神經系統(myocardialsympaticnervesystem)、白三烯受體(例如在白細胞上)、尿激酶纖維蛋白溶酶原激活劑受體(uPAR)、葉酸鹽受體、細胞凋亡標志物、(抗)血管生成標志物、胃泌素受體、多巴胺能系統、serotonergic系統、GABA能系統(GABAergicsystem)、腎上腺素反應系統、膽堿能系統、阿片受體、GPIIb/IIIa受體和其它血栓相關受體、纖維蛋白、降鈣素受體、促吞噬素受體、整合素受體、VEGF/EGF受體、EGF、基質金屬蛋白酶(MMP)、P/E/L-選擇素受體、LDL受體、P-糖蛋白、神經降壓素受體、神經肽受體、P物質受體、NK受體、CCK受體、σ受體、白細胞介素受體、單純皰疹病毒酪氨酸激酶、人酪氨酸激酶。根據本發明的一個特定實施方案,所述初級靶是蛋白質例如受體。作為可選選擇,所述初級靶可以是代謝途徑,其在疾病例如感染或癌癥期間表達上調,例如DNA合成、蛋白質合成、膜合成和碳水化合物攝取。在患病組織中,上述標志物能夠不同于健康組織并提供早期檢測、特異性診斷和治療特別是靶向的治療的獨特可能。預靶向探針預靶向探針包含能夠與感興趣的初級靶結合的部分。靶向部分典型地是對細胞表面靶(例如膜受體)、結構蛋白(例如淀粉樣斑)或細胞內靶(例如RNA、DNA、酶、細胞信號通路)具有親和性的結構。這些部分可以是抗體(片段)、蛋白質、適體、寡肽、寡核苷酸、寡糖以及肽、擬肽和已知在特定的疾病或障礙處積累的有機藥物化合物。本文描述了用于本發明試劑盒的適合初級靶向部分的特定實施方案,并包括受體結合性肽和抗體。本發明的一個特定實施方案涉及小靶向部分例如肽的使用,從而獲得可透過細胞的靶向探針。本發明中使用的“初級靶向部分”涉及結合到初級靶的靶向探針部分。初級靶向部分的特定例子是結合到受體的肽或蛋白質。初級靶向部分的其它例子是結合到細胞化合物的抗體或其片段。抗體可以針對非蛋白質化合物和蛋白質或肽生成。其它初級靶向部分可以由適體、寡肽、寡核苷酸、寡糖、以及擬肽和有機藥物化合物構成。初級靶向部分優選以高特異性、高親和性、任選地甚至共價結合,并且與所述初級靶的結合優選在體內是穩定的。為了使得特異性靶向以上列舉的初級靶,所述靶向探針的初級靶向部分可以含有化合物,其包括但不限于:抗體、抗體片段例如Fab2、Fab、scFV、雙價抗體(diabodies)、聚合物(依靠EPR效應的腫瘤靶向)、蛋白質、肽例如奧曲肽和衍生物、VIP、MSH、LHRH、趨化肽、鈴蟾肽、彈性蛋白、肽類似物、碳水化合物、單糖、多糖、病毒、全細胞(wholecells)、噬菌體、藥物、化療劑、受體激動劑和拮抗劑、細胞活素、荷爾蒙、類固醇。在本發明的內容中設想的有機化合物的例子是或衍生自:雌激素類例如雌二醇、雄激素類、孕激素類、皮質類固醇類、紫杉醇、足葉乙甙、doxorubricin、甲氨喋呤、葉酸和膽固醇。根據本發明的一個特定實施方案,所述初級靶是受體,并且適合的初級靶向部分包括但不限于此類受體的配體或仍結合到該受體的配體部分,例如在受體結合性蛋白質配體的情況下受體結合性肽。蛋白質性質的初級靶向部分的其他例子包括干擾素,例如α、β和γ干擾素,白細胞介素和蛋白質生長因子,例如腫瘤生長因子,例如α、β腫瘤生長因子,血小板衍生生長因子(PDGF),uPAR靶向蛋白,載脂蛋白,LDL,膜聯蛋白V,內皮抑素和血管生長抑制素。初級靶向部分的可選擇的例子包括DNA、RNA、PNA和LNA,其例如與所述初級靶互補。根據本發明的一個特定實施方案,使用了小的親脂性初級靶向部分,其能夠結合到細胞內初級靶。根據本發明的一個進一步的特定實施方案,選擇所述初級靶和初級靶向部分從而導致特異性的或增加的組織或疾病的靶向,所述組織或疾病例如癌癥、炎癥、感染、心血管疾病例如血栓、動脈粥樣硬化病變、缺氧位置例如中風、腫瘤、心血管障礙、腦失調、細胞凋亡、血管生成、器官和報告基因/酶。這可以通過選擇具有組織、細胞或疾病特異性表達的初級靶實現。例如,膜葉酸受體介導葉酸鹽及其類似物例如甲氨喋呤的細胞內積累。正常組織內表達是有限的,但受體在各種腫瘤細胞類型中被過度表達。根據一個實施方案,所述預靶向探針和所述效應物探針可以是多體化合物(multimericcompounds),其包含多個初級和/或次級靶和/或靶向部分。這些多體化合物可以是聚合物、樹枝狀聚合物、脂質體、聚合物顆粒或其它聚合物結構。對于放大檢測信號特別感興趣的是具有多于一個次級靶的靶向探針,其使得能夠結合數個效應物探針。所述預靶向探針進一步含有上述第一生物正交反應性基團。該基團充當“次級靶”,即作為提供用于所述逆Diels-Alder耦合化學的第一反應參與者的靶向探針部分。如上所述,所述次級靶可以是所述耦合反應的任一參與者。即,在一個實施方案中,其是缺電子四嗪。在另一實施方案中,其是上述式(1)的直立取代的TCO。在所述預靶向探針中,所述初級靶向部分和所述第一生物正交反應性基團可以彼此直接相連。它們還可以通過連接體彼此結合,此外它們可以都與初級靶向支架(scaffold)例如生物聚合物如多肽連接。即在最簡單的情況下,所述連接體部分是鍵。適合的連接體部分進一步包括,但不限于,具有從2到200,特別是3到113和優選5-50個重復單元的聚乙二醇(PEG)鏈。通過調節PEG鏈長,能夠影響所述探針在生理體系中的循環時間。這對于預靶向探針是特別相關的(因為將所述初級靶向部分連接到所述初級靶的初始靶向步驟可能涉及相對慢的過程,需要相對長的循環時間)。連接體部分任選地包括生物聚合物片段,例如寡-或多肽或聚交酯(polylactides)。應該理解,本發明包括其中所述二烯和所述親雙烯體與所述預靶向或效應物探針任一連接的任何可想到的方式。實施對這些探針的結合的方法,例如通過反應性氨基酸例如賴氨酸或半胱氨酸,已為本領域技術人員所知曉。效應物探針效應物探針包含能夠提供想要的診斷、成像和/或治療效果的效應物部分。所述效應物探針進一步包含次級靶向部分。所述次級靶向部分涉及形成用于可用的次級靶(即所述預靶向探針中包含的一個或多個生物正交反應性基團)的反應參與者的效應物探針部分。將理解,在所述次級靶是如上定義的式(1)的直立取代的TCO的情況下,所述次級靶向部分將是二烯例如四嗪,反之亦然。所述效應物部分可以是例如可檢測的標記物。這里使用的“可檢測標記物”涉及使得例如當存在于細胞、組織或生物體中時所述探針能夠被檢測的效應物探針部分。在本發明的內容中,所設想的可檢測標記物的一種類型是對比提供劑。在本發明的內容中設想了不同類型的可檢測標記物并在下文加以描述。因此,根據本發明的一個特定實施方案,本發明的預靶向試劑盒和方法被用于成像,特別是醫學成像。為了識別所述初級靶,使用包含一種或多種可檢測標記物的成像探針作為所述效應物探針。所述成像探針的可檢測標記物的特定例子是用于常規成像體系的對比提供部分例如可MRI-成像的結構、自旋標記物、光學標記物、超聲響應結構、X射線響應部分、放射性核素、(生物)發光和FRET類型染料。在本發明的內容中所設想的示例性的可檢測標記物包括但不一定局限于熒光分子(例如自發熒光分子(autofluorescentmolecules)、與試劑接觸時發熒光的分子等)、放射標記物;生物素(例如將要經由用抗生物素蛋白結合生物素進行檢測的生物素);熒光標簽,用于MRI的成像結構,包括順磁性金屬,成像試劑,例如在U.S.Pat.No.4,741,900和5,326,856中描述的那些)等。用于成像的放射性核素可以例如是選自由以下組成的組的同位素:3H、11C、13N、15O、18F、19F、51Cr、52Fe、52Mn、55Co、60Cu、61Cu、62Zn、62Cu、63Zn、64Cu、66Ga、67Ga、68Ga、70As、71As、72As、74As、75Se、75Br、76Br、77Br、8OBr、82Br、82Rb、86Y、88Y、89Sr、89Zr、97Ru、99Tc、110In、111In、113In、114In、117Sn、120I、122Xe、123I、124I、125I、166Ho、167Tm、169Yb、193Pt、195Pt、201Tl、和203Pb。在某些應用中也可以將其它元素和同位素例如被用于治療的元素或同位素用于成像。所述可MRI成像的部分可以是例如順磁性離子或超順磁性顆粒。所述順磁性離子可以是選自由以下組成的組的元素:Gd、Fe、Mn、Cr、Co、Ni、Cu、Pr、Nd、Yb、Tb、Dy、Ho、Er、Sm、Eu、Ti、Pa、La、Sc、V、Mo、Ru、Ce、Dy、Tl。所述超聲響應部分可以包含微泡,其殼由磷脂和/或(可生物降解的)聚合物、和/或人血清白蛋白構成。所述微泡可以用氟化氣體或液體填充。所述X射線響應部分包括但不限于碘、鋇、硫酸鋇、gastrografin或可以包含用碘化合物和/或硫酸鋇填充的囊泡、脂質體或聚合物膠囊。此外,本發明的內容中所設想的可檢測標記物還包括能夠通過抗體結合進行檢測的肽或多肽,例如通過結合可檢測的經標記的抗體或通過經由夾心式檢驗檢測結合的抗體。在一個實施方案中,所述可檢測標記物是小尺寸有機PET和SPECT標記物,例如18F、11C或123I。由于它們的小尺寸,有機PET或SPECT標記物完美地適合用于監控細胞內的事件,因為它們一般不會很大地影響靶向器件的性能并且特別是其膜輸送。包含PET標記物和作為次級靶向部分的任一逆Diels-Alder活性部分的成像探針是親脂性的并且能夠被動地擴散進和擴散出細胞直到它找到它的結合參與者。而且,兩種組分都不妨礙越過血腦屏障并從而使得能夠在腦內區域成像。當所述效應物探針打算包含基于金屬例如用于MRI對比增強的鑭系金屬(例如Gd)的可檢測標記物時,其優選以螯合物的形式提供。在這種情況下,所述效應物探針優選包含能夠與此類金屬形成配位絡合物的結構部分。關于此的一個好例子是衍生自1,4,7,10-四氮雜環十二烷-1,4,7,10-四乙酸(H4dota)和1,4,7,10-四氮雜環十二烷-α,α’,α,”α’”-四甲基-1,4,7,10-四乙酸(H4dotma)的大環鑭系元素(III)螯合物。所述效應物部分還可以是治療部分例如藥學活性化合物。本文提供了藥學活性化合物的例子。治療探針還可以任選地包含可檢測標記物。因此,根據另一實施方案,本發明的預靶向試劑盒和方法被用于靶向的治療。這通過利用含有次級靶向部分和一個或多個藥學活性試劑(即藥物或用于放射療法的放射性同位素)的效應物探針實現。用于靶向的藥物遞送情況的適合藥物已為本領域所知。任選地,所述治療探針還可以包括可檢測的標記物,例如一種或多種成像劑。用于治療的放射性核素可以是例如選自由以下組成的組的同位素:24Na、32P、33P、47Sc、59Fe、67Cu、76As、77As、80Br、82Br、89Sr、90Nb、90Y、103Ru、105Rh、109Pd、111Ag、111In、121Sn、127Te、131I、140La、141Ce、142Pr、143Pr、144Pr、149Pm、149Tb、151Pm、153Sm、159Gd、161Tb、165Dy、166Dy、166Ho、169Er、172Tm、175Yb、177Lu、186Re、188Re、198Au、199Au、211At、211Bi、212Bi、212Pb、213Bi、214Bi、223Ra和225Ac。作為選擇,所述治療探針中的藥物選自用于光動力療法的感光劑(sensitizers)。作為選擇,所述治療探針包含結合到體內治療實體,例如T細胞、自然殺傷細胞或其它內源性結構例如蛋白質,的識別部分。在所述效應物探針中,所述次級靶向部分即所述第二生物正交反應性基團和所述效應物部分可以直接彼此連接。它們還可以通過連接體彼此結合,此外它們可以都與次級靶向支架連接。所述連接體可以獨立地選自如上所述的相同部分,例如聚乙二醇。所述次級靶向支架可以是例如生物聚合物如多肽。本發明還涉及利用逆Diels-Alder反應的預靶向方法。在這里,將包含初級靶向部分(例如抗體和抗體片段或受體結合性肽)的預靶向探針注入到受試者中,其中所述預靶向探針分別用適合的二烯,優選根據上述式(2)-(5)任意一個的化合物,或用根據上述式(1)的環辛烯官能化。在結合到靶(例如原發或轉移性腫瘤病變、動脈粥樣硬化斑塊、梗死區域、炎癥或感染位置等)并從循環系統和從非靶組織(例如血液、肝臟、脾臟、腎臟等)清除之后,注入效應物探針,其含有次級靶向部分例如分別攜帶E-環辛烯或四嗪衍生物(即存在于所述預靶向探針中的生物正交反應性基團的反應性對應物)和藥物或可成像標記物。所述效應物探針結合到所述初級靶向部分并提供高對比或選擇性治療所述疾病位點。本發明還涉及一般的代謝途徑的靶向,所述一般的代謝途徑在疾病(例如感染或癌癥)期間表達上調如DNA、蛋白質和膜合成以及碳水化合物攝取。適合的探針包含被二烯或親雙烯體標記的氨基酸、糖、核酸和膽堿,本領域中目前使用的代謝示蹤劑類似物,[11C]-蛋氨酸、[18F]-氟脫氧葡萄糖(FDG)、脫氧-[18F]-氟胸苷(FLT)和[11C]-膽堿。具有高代謝或增殖的細胞具有對這些構建單元(buildingblock)的較高攝取。在該方法中,例如四嗪或E-環辛烯衍生物進入這些或其它通路并在細胞內和/或細胞上積累。在充分積聚并清除游離探針后,將經可檢測地標記的或攜帶藥物的(細胞可透過)的四嗪探針或E-環辛烯探針(或攜帶根據本發明的其它二烯/親雙烯體的探針)送入以分別結合積累的E-環辛烯或四嗪代謝物。作為優于普通FDG(氟18氟脫氧葡萄糖)類型成像的一個益處,可以獲得足夠的時間使得能夠在送入放射性之前高度積聚所述靶向部分,從而增大靶對非靶比。作為選擇,可以靶向疾病特異性的代謝途徑和/或代謝物。本發明還涉及細胞內靶的預靶向。由于它們的小尺寸,有機PET標記物(18F,11C)非常適合用于監控細胞內事件,因為它們通常不會很大地影響所述靶向器件的性能,特別是其膜輸送(和大的和極性的放射金屬螯合物結構綴合物相反)。盡管在本發明中使用的被取代的四嗪部分和所述E-環辛烯不一定是小的,但是它們是相對非極性的,并且能夠用于蛋白質、mRNA、信號通路等的分子內成像。所述次級(例如PET標記的)取代的四嗪部分或E-環辛烯探針(即所述效應物探針)能夠被動地擴散進和擴散出細胞直到它找到它的結合參與者或經歷活性攝取機理。這些性能還允許將逆Diels-Alder反應用于腦內預靶向,因為兩種組分都不妨礙越過血腦屏障。本發明還涉及預靶向的信號放大和/或多價裝配。至少一個初級靶向器件與含有多個四嗪部分的樹枝狀聚合物(dendrimer)、聚合物或納米顆粒結合。在受體結合后,注入與用于核成像(例如放射金屬螯合物、放射性鹵素等)或MRI(例如Gd螯合物)的一個或多個對比部分結合的(一種或多種)環辛烯。隨后的逆Diels-Alder反應導致在靶組織處MRI對比劑的高濃度。此外,在靶位點的多價將增大與所述直立取代的TCO效應物綴合物的反應動力學,提供例如MRI對比劑的有效靶積累。當然,所述直立取代的TCO也可以用于所述靶向器件綴合物中而所述四嗪(或本發明的其它二烯)連接到報告物(reporter)。連接途徑和試劑盒本發明進一步涉及逆Diels-Alder反應作為用于將成像劑和藥物連接到靶向結構例如肽的途徑的用途。所述效應物可以包含有機PET或SPECT核素標記的輔基、用于PET/SPECT/MRI的金屬絡合物和用于超聲成像的微泡、用于光學成像的熒光團以及用于放射療法的α和β輻射源和通常細胞毒素抗癌劑。所述成像/治療劑可以用側基四嗪或其它適合的二烯部分官能化和用直立取代的TCO衍生物將靶向基團官能化,反之亦然。本發明的途徑對用于核成像和放射療法的試劑特別有利:考慮到放射性核素的衰變,作為預靶向步驟實施最耗時的步驟(受試者體內的實際靶向)是有益的。根據本發明,選擇直立取代的TCO,以獲得上述非常快速的逆Diels-Alder化學用于次級靶向,使得能夠使用多種放射性核素,包括與現有方法相比使用壽命更短的那些。直立取代的TCO官能化的效應物探針和適合的二烯例如攜帶四嗪的預靶向探針能夠在極低濃度在體內耦合而不需要效應物部分(例如所述放射性核素)的持續血液循環。將理解,這對與二烯特別是四嗪官能化的效應物探針結合的帶有直立取代的TCO的預靶向探針同樣適用。而且,所述反應性基團有利地是穩定的,并因此展現了較長壽命的反應性而不過于容易地發生副反應。將理解,以上提供了益處例如最小化對病人的放射劑量。而且,其導致能夠利用PET即正電子發射斷層成像劑代替SPECT即單光子發射計算機斷層成像劑(singlephotonemissioncomputerizedtomographyagents)。而且,增加的反應性使得能夠在體內以較低的濃度應用。本發明特別適合用于多模式成像,任選地使用不同的成像劑以使相同的靶可視化。作為選擇,所述成像探針包含至少兩種不同的標記物以使得能夠實現多模式成像。本發明的改善的[4+2]逆Diels-Alder化學在分子成像中的應用向所有類型和尺寸的靶向結構打開了預靶向之門。這允許細胞內和代謝成像以從可通過預靶向積聚獲得的高靶積累和低背景受益。同樣,對于較小的和更多種多樣的靶向器件,預靶向的信號放大方案例如多四嗪和/或多樹枝狀聚合物或脂質體變得可行。由于所述反應參與者是非生物的和生物正交的,因此使用利用直立取代的TCO作為上述親雙烯體的[4+2]逆Diels-Alder反應的預靶向不受內源性競爭和代謝/分解妨礙,并提供穩定的共價鍵。選擇靶代謝途徑,以及相應的四嗪-代謝物衍生物,憑借其與正常細胞相比在例如腫瘤細胞中的高通量,提供了在細胞中或在靶細胞表面上高密度人工四嗪受體或其它化學柄的裝配,避免有時可能處于低水平的內源細胞表面受體的使用。本發明的進一步的特定實施方案涉及包含代謝前體和成像探針,更特別地是包含可檢測標記物的成像探針的試劑盒,所述可檢測標記物是用于常規成像體系的對比劑。此類可檢測標記物可以是但不限于選自由可MRI成像的結構、自旋標記物、光學標記物、超聲響應試劑、X-射線響應試劑、放射性核素和FRET類染料組成的組的標記物。在本發明的一個特定實施方案中,使用了報告物探針。這種報告物探針可以是酶的底物,更特別地是對于所述細胞不是內源性的,但已經通過基因療法或使用外來試劑感染而引入的酶。在本文中涉及細胞或組織內的基因的非內源性用于表示所述基因在所述細胞或組織內并不天然存在和/或表達。作為選擇,此類報告物探針是通過受體或泵的方式引入到細胞中的分子,其可以是內源性的或通過基因療法或使用外來試劑感染引入到細胞內。作為選擇,所述報告物探針是對細胞或組織環境內的某些(變化)條件起反應的分子。本發明還包括用于上述試劑盒中的試劑。一種此類試劑是包含初級靶向部分和生物正交反應性基團的預靶向劑,其中所述生物正交反應性基團是[4+2]逆Diels-Alder反應的反應參與者。特定反應參與者在上文描述,即通常是上述缺電子四嗪或其它適合的二烯,或是根據本發明的直立取代的環辛烯。本發明還涉及這些試劑在靶向的醫學成像或靶向的治療中的用途,和涉及用于這種方法中的所述試劑。特別地,本發明涉及這些試劑在預靶向方法中的這些用途,和涉及在這種方法中使用的這些試劑。另一種此類試劑是包含可檢測標記物和生物正交反應性基團的成像探針,其中所述生物正交反應性基團是[4+2]逆Diels-Alder反應的反應參與者。本發明還涉及包含可檢測標記物和生物正交反應性基團的成像探針,其中所述生物正交反應性基團是[4+2]逆Diels-Alder反應的反應參與者。本發明進一步涉及包含藥學活性化合物和生物正交反應性基團的治療探針,其中所述生物正交反應性基團是[4+2]逆Diels-Alder反應的反應參與者。本發明的一部分還是預靶向方法,包括將如上所述的預靶向試劑給予受試者并使所述試劑在受試者的體系中循環有效實現所述初級靶向部分對初級靶的結合的一段時間,接著從身體清除未結合的試劑。用于其的典型時間為12到96小時,特別地為約48小時。此外,本發明提供了成像方法,包括如上所述實施預靶向方法,接著給予同樣根據本發明的成像探針,其中所述預靶向試劑中和所述成像探針中的生物正交反應性基團一起形成[4+2]逆Diels-Alder反應的反應參與者。類似地,本發明提供了受試者中靶向的醫學治療方法,其包括如上所述實施預靶向方法,接著給予同樣根據本發明的治療探針,其中所述預靶向試劑中和所述成像探針中的生物正交反應性基團一起形成[4+2]逆Diels-Alder反應的反應參與者。本發明還涉及用于如上所述的成像或治療方法的上述預靶向試劑。總結而言,在逆Diels-Alder化學的基礎上,生物正交預靶向的分子成像和治療用于給病人帶來極大益處。一方面,其用于承擔獲得靶組織例如癌癥和心血管病變的優良圖像。另一方面,能夠大大減少源自放射性化合物和通常潛在毒性藥物給予的固有副作用,同時增加到達患病組織的有效劑量。此外,其將大大擴展引起疾病的可追蹤分子事件的收集。特別地,這種技術能夠允許使用遠離血管的靶組織并將促進信息豐富的細胞內環境的成像。將參考以下非限定性實施例和隨附的非限定性附圖說明本發明。實施例材料所有試劑和溶劑都從商業來源獲得(從Sigma-Aldrich、Acros、ABCR、Invitrogen和Merck獲得試劑,從Biosolve、Merck和CambridgeIsotopeLaboratories獲得普通和氘代溶劑),并除非另外說明都不經進一步的純化即使用。1-氨基-3,6,9,12,15,18,21,24,27,30,33,36-十二氧雜三十九烷-39-酸(21)從Polypure獲得。[111In]氯化銦和[125I]碘化鈉溶液購自PerkinElmer。通過milli-Q水過濾系統(Millipore)蒸餾和去離子化水(18MΩcm)。用Chelex-100樹脂(BioRadLaboratories)處理標記緩沖液過夜,然后經過0.22μm過濾并在4℃貯存。Indogen碘化管、用于二喹啉甲酸(BCA)檢驗的試劑盒、gelcode藍色蛋白染色溶液和Zeba脫鹽離心柱(spincolumns)(40kDaMW截止,0.5-2mL)購自PierceProteinResearch(ThermoFisherScientific)。用于制備磷酸鹽緩沖鹽水(PBS)pH7.4的片劑從Calbiochem(Merck)獲得。AmiconUltra-4和Ultra-15離心過濾器單元(50kDaMW截至)購自Millipore。小鼠血清購自InnovativeResearch。四嗪28的合成和放射標記如Rossin等,AngewChemIntEd2010,49,3375中描述的那樣實施。方法使用BrukerDPX300波譜儀或BrukerAvance600波譜儀在CDCl3或[D6]DMSO中記錄NMR譜。使用DEPT脈沖序列區分13C-NMR多重性(q=四重、t=三重、s=二重和p=一重(primary))。在AgilentESI-TOF質譜儀上記錄了高分辨率ESI質譜(HRMS),以陽離子模式測量。在CombiflashCompanion設備(TeledyneIsco)上使用SiliCycle硅膠柱實施制備柱色譜。使用裝配有C18Zorbax柱(21.2×150mm,5μm顆粒)的Agilent1200儀器,應用含有0.1%TFA的水和MeCN梯度,實施制備HPLC。在裝配有Gabi放射性探測器(Raytest)的Agilent1100系統上實施分析放射-HPLC。將試樣加載在AgilentEclipseXDB-C18柱(4.6×150mm,5μm顆粒)上,其用含有0.1%TFA的MeCN在水中的線性梯度以1mL/min洗脫(10%MeCN2min,接著在11min內增大到45%MeCN)。將UV波長預設在254nm。在裝配有Gabi放射性探測器的Agilent1200系統上實施體積排阻(SEC)HPLC。將試樣加載在BioSep-SEC-S2000柱(300×7.8mm,5μm顆粒,Phenomenex)上并用20mM磷酸鹽、150mMNaCl、pH6.8以1mL/min洗脫。UV波長預設在260和280nm。使用用在0.9%NaCl水溶液中的200mMEDTA洗脫并在磷光體成像儀(FLA-7000,Fujifilm)上成像的ITLC-SG條(Pall)通過放射-TLC測定了111In和177Lu標記產率。在這些條件下,游離111In和177Lu以Rf=0.9遷移,而111In/177Lu-四嗪保留在原位。125I-標記產率也通過放射-TLC測定,使用用1:1MeOH/醋酸乙酯混合物洗脫并在磷光體成像儀上成像的ITLC-SG條。在這些條件下,游離125I和125I-SHPP以Rf=0.9遷移,而125I-mAbs保留在原位。在Phastgel系統上分別使用IEF-3-9凝膠和7.5%PAGE均勻凝膠(homogeneousgels)(GEHealthcareLifeSciences)實施等電聚焦(IEF)分析和SDS-PAGE。IEF校準溶液(寬PI,pH3-10)購自GEHealthcare和蛋白質MW標準溶液(PrecisionPlus雙色標準)購自BioRad。在電泳時,用gelcode藍將所述凝膠染色2小時,在水中脫色過夜然后用常規平板掃描儀數字化。用NanoDrop1000分光光度計(280nm處的吸光率;ThermoFisherScientific)或采用BCA測試測定CC49溶液的濃度。LS174T腫瘤模型(tumormodel)。人克隆癌細胞系LS174T從ATCC獲得并保存在以10%熱滅活的胎牛血清(Gibco)、青霉素(100U/mL)、鏈霉素(100μg/mL)和2mMGlutamax補充的Eagle’s最小必需培養基(Sigma)中。用100μL無菌PBS中的5×106細胞對裸雌性Balb/C小鼠(20-25克體重,CharlesRiverLaboratories)進行皮下接種。實施例1如圖3a中所示,作為將四嗪衍生的部分連接到抗體的一個實例,制備了分子1(見圖5)。相應的探針2(衍生自E-環辛烯)的一個實例在圖6中示出。兩種分子都含有PEG鏈。分子1包含用于將所述分子與抗體中存在的氨基基團耦合的N-羥基琥珀酰亞胺基部分。2中DOTA衍生的部分可以用于攜帶稀土金屬離子例如用于MR成像的Gd或用于核成像和治療(SPECT)的Lu-177。圖5中示出了1的合成。根據Blackman等(Blackman,ML;Royzen,M;Fox,JM,JournalofTheAmericanChemicalSociety,2008,130(41),13518-19)制備了起始的四嗪衍生的分子5。通過與戊二酸酐反應將其轉化成酸6,接著形成它的N-羥基琥珀酰亞胺基酯7。該N-羥基琥珀酰亞胺基酯用于通過與商購獲得(IRISbiochem)的PEG衍生物8反應形成酸9,其進而被轉化成它的N-羥基琥珀酰亞胺基酯1。圖6中示出了2的合成。根據Yap等(Yap,GPA;Royzen,M;Fox,JM,JournalofTheAmericanChemicalSociety,2008,130(12),3760–61)制備了(E)-環辛-4-烯醇(10)。其在商購獲得的(Aldrich)異氰酸酯衍生物11的幫助下被轉化成酯12,接著皂化成酸13。使由13形成的N-羥基琥珀酰亞胺基酯14與衍生自DOTA和PEG的胺18反應,以形成終產物2。使17脫保護后制備DOTA衍生物18,所述17進而由DOTA衍生物15和PEG衍生物16(都可商購獲得)(分別來自Macrocyclics和IRISBiotech)制備。實施例2本實施例闡述了實施例1的分子的相反對,圖7中示出了用于結合到抗體后形成所述預靶向部分的所述E-環辛烯衍生物3。能夠用作圖3b中示出的效應物探針的四嗪/DOTA衍生的探針4在圖8中示出。E-環辛烯衍生物3通過如下方式形成:商購獲得(IRISbiochem)的PEG衍生物8(也見圖5)與N-羥基琥珀酰亞胺基酯14反應以形成酸19,接著形成源自這種酸的N-羥基琥珀酰亞胺基衍生物(圖7)。圖8中示出了所述四嗪/DOTA衍生的探針4的合成。所述探針通過DOTA和PEG衍生的胺18(見圖6)與N-羥基琥珀酰亞胺基酯7(見圖5)的反應制備。實施例3本實施例示于圖9中,其提供了用于合成(E)-2,5-二氧代吡咯烷-1-基1-(4-((環辛-4-烯-1-基氧基)甲基)苯基)-1-氧代-5,8,11,14,17,20,23,26,29,32,35,38-十二氧雜-2-氮雜四十一烷-41-酸酯(TCO-O-PEG10-N-羥基琥珀酰亞胺(NHS),主要和次要異構體分別是23a和23b)的方案。標記為(數字)a的化合物代表E-主要和標記為(數字)b的化合物代表E-次要。(E-主要)-2,5-二氧代吡咯烷-1-基4-((環辛-4-烯基氧基)甲基)苯甲酸酯(20a)。根據文獻過程(M.Royzen,G.P.A.Yap,J.M.Fox,J.Am.Chem.Soc.2008,130,3760)合成了(E)-環辛-4-烯醇(10a,主要異構體,含有約13%所述Z-異構體)。將60%氫化鈉分散體(1.8g,45mmol)加入冰浴冷卻的10a(1.70g,13.5mmol)在60mLDMF中的溶液中。在室溫攪拌4小時后,將4-溴甲基苯甲酸(3.85g,17.9mmol)分份加入并在室溫將懸浮液攪拌過夜。將混合物倒入水(100mL)中,加入叔丁基甲基醚(100mL),接著加入37%鹽酸(5mL)。分離后,用叔丁基甲基醚(2×100mL)提取水層。用水(25mL)洗滌合并的有機層,用MgSO4干燥并蒸發。用4:1己烷/乙酸乙酯使剩余物通過薄硅膠層。將蒸發后獲得的剩余物在70℃溶解在庚烷(50mL)中,然后冷卻,提供19a。將所述產物溶解在二氯甲烷(40mL)中,加入N-羥基琥珀酰亞胺(0.57g,4.9mmol),將混合物在冰浴中冷卻,接著加入N,N’-二環己基碳二亞胺(1.03g,4.99mmol)。30分鐘后除去冰浴并在室溫攪拌所述反應混合物18小時。過濾并蒸發后,通過柱色譜在硅膠上使用乙酸乙酯在庚烷(0-15%)中的梯度純化剩余物。接著,將所述剩余物溶解在叔丁基甲基醚(20mL)中并倒入庚烷(50mL)中,得到白色固體形式的20a(1.42g,29%)。(E-主要)-2,5-二氧代吡咯烷-1-基1-(4-((環辛-4-烯-1-基氧基)甲基)苯基)-1-氧代-5,8,11,14,17,20,23,26,29,32,35,38-十二氧雜-2-氮雜四十一烷-41-酸酯(23a)。將20a(100mg,0.280mmol)在二氯甲烷(2mL)中的溶液滴加入在冰浴中攪拌的21(175mg,0.283mmol)和三乙胺(290μL,2.08mmol)在二氯甲烷(2mL)中的溶液中。將反應混合物在室溫攪拌16小時。將蒸發后所獲得的粗中間體22a溶解在二氯甲烷(5mL)中并在冰浴中冷卻。加入雙(2,5-二氧代吡咯烷-1-基)碳酸酯(170mg,0.664mmol)和吡啶(28μL,0.35mmol),并在室溫將反應混合物攪拌3小時。過濾混合物并蒸發,通過柱色譜在硅膠上使用甲醇在二氯甲烷(5-10%)中的梯度純化產物,得到粘性油形式的23a(119mg,39%)。(E-次要)-2,5-二氧代吡咯烷-1-基4-((環辛-4-烯基氧基)甲基)苯甲酸酯(20b)。根據上述文獻過程合成(E)-環辛-4-烯醇(10b,次要異構體)。將60%氫化鈉分散體(2.1g,53mmol)加入冰浴冷卻的10b(2.53g,20.1mmol)在50mL四氫呋喃中的溶液中。在室溫攪拌4小時后,將混合物再次冷卻并用5分鐘的時間分份加入4-溴甲基苯甲酸(4.53g,21.1mmol)。加入25mL四氫呋喃并在將所述懸濁液在室溫攪拌4天。加入冰,接著加入12.0g檸檬酸。用150mL叔丁基甲基醚萃取所述混合物兩次。有機層用25mL水洗滌、干燥并蒸發。通過使用80g硅膠和含有逐步增加的量的醋酸乙酯的庚烷作為洗脫液的色譜法純化剩余物。合并產物級分(產物和起始醇之間未完全分離)并從約30mL庚烷重結晶(冷卻到-15℃),得到白色固體形式的19b(0.86g,17%)。將其溶解在40mL二氯甲烷中。加入N-羥基琥珀酰亞胺(0.48g,4.17mmol)并在冰中冷卻混合物。加入N,N’-二環己基碳二亞胺(0.80g,3.88mmol)并將混合物在冰中攪拌30分鐘,然后在室溫攪拌18小時。通過過濾、用二氯甲烷洗滌、旋轉蒸發以及在25g硅膠上用庚烷-醋酸乙酯作為洗脫液的色譜法提供了產物。將蒸發的產物級分與75mL叔丁基甲基醚混合并將混合物加溫到60℃以提供溶液。將所述溶液濃縮到20mL。逐步加入50mL庚烷獲得所述產物的沉淀。通過過濾收集和用庚烷洗滌提供1.05g20b(15%)。(E-次要)-2,5-二氧代吡咯烷-1-基1-(4-((環辛-4-烯-1-基氧基)甲基)苯基-1-氧代-5,8,11,14,17,20,23,26,29,32,35,38-十二氧雜-2-氮雜四十一烷-41-酸酯(23b)。該化合物以與23a相似的方式從20b開始制備。以93%的產率獲得粘性油形式的23b。實施例4本實施例示于圖10中,其給出了用于合成(E)-2,5-二氧代吡咯烷-1-基1-(4-(((環辛-4-烯-1-基氧基)羰基)氨基)苯基)-1-氧代-5,8,11,14,17,20,23,26,29,32,35,38-十二氧雜-2-氮雜四十一烷-41-酸酯的反應方案。標記為(數字)a的化合物代表E-主要和標記為(數字)b的化合物代表E-次要。(E-主要)-2,5-二氧代吡咯烷-1-基4-(((環辛-4-烯-1-基氧基)羰基)氨基)苯甲酸酯14a。將N,N’-二環己基碳二亞胺(9.50g,0.046mol)分份加入在冰中冷卻的4-氨基苯甲酸(5.84g,0.043mol)、N-羥基琥珀酰亞胺(5.0g,0.044mmol)和60mL異丙醇的混合物中。在室溫攪拌16小時后過濾懸濁液并用60mL異丙醇將所獲得的固體再攪拌一小時。將過濾并真空下干燥后獲得的粗固體24與100mL二氯甲烷混合并在冰中冷卻。加入20%碳酰氯在甲苯中的溶液(26mL,49.4mmoL)并攪拌30分鐘后獲得溶液。將蒸發后獲得的固體用100mL甲苯洗滌兩次并在高真空下干燥。獲得4.03g白色固體形式的25(36%)。將所述固體分份加入10a(1.40g,11.1mmol)在35mL二氯甲烷中的溶液中。將所述懸浮液攪拌過夜,并然后在40℃加溫2小時。在二氧化硅上用二氯甲烷洗脫,接著從甲苯重結晶后,獲得1.77g固體形式的14a(41%)。所述產物包含約12%的Z-異構體,其不能通過色譜法或結晶除去。(E-主要)-2,5-二氧代吡咯烷-1-基1-(4-(((環辛-4-烯-1-基氧基)羰基)氨基)苯基)-1-氧代-5,8,11,14,17,20,23,26,29,32,35,38-十二氧雜-2-氮雜四十一烷-41-酸酯27a。將14a(100mg,0.26mmol)在二氯甲烷(2mL)中的溶液滴加加入在冰浴中攪拌的21(160mg,0.26mmol)和三乙胺(400μL,2.9mmol)在二氯甲烷(2mL)中的溶液中。將所述反應混合物在室溫攪拌16小時。將蒸發后獲得的粗中間體26a溶解在二氯甲烷(5mL)中,并在冰浴中冷卻。加入雙(2,5-二氧代吡咯烷-1-基)碳酸酯(89mg,0.35mmol)和吡啶(28μL,0.35mmol)并將所述反應混合物在室溫攪拌16小時。過濾混合物,用3mL水提取3次和用3mL鹽水提取一次。用硫酸鎂干燥并蒸發后獲得1.23g粘性油形式的27a(47%)。(E-次要)-2,5-二氧代吡咯烷-1-基4-(((環辛-4-烯-1-基氧基)羰基)氨基)苯甲酸酯14b。以與制備14a相似的方式但從10b開始以32%產率獲得該化合物。由于獲得了純凈的10b,因此該產物不含所述Z-異構體。(E-次要)-2,5-二氧代吡咯烷-1-基1-(4-(((環辛-4-烯-1-基氧基)羰基)氨基)苯基)-1-氧代-5,8,11,14,17,20,23,26,29,32,35,38-十二氧雜-2-氮雜四十一烷-41-酸酯27b:以與制備27a相似的方式,但從14b開始以89%產率獲得該化合物。實施例5四嗪放射標記將DOTA-結合的四嗪(28;圖12,在Rossin等AngewChemIntEd2010,49,3375中描述)溶解(1mg/mL)在0.2M醋酸銨pH7.0中并且使用前在-80℃貯存。將等分量的28與適合量的[111In]氯化銦或[177Lu]氯化镥組合并在37℃在輕柔攪拌下培育10分鐘。然后加入5μL10mmDTPA并將所述溶液再額外培育5分鐘。通過對于所述四嗪加入0.9摩爾當量的InCl3或LuCl3實施載體加入的標記反應。典型地,采用該方法獲得了大于98%的定量標記產率和放射化學純度。與反式環辛烯(TCO)NHS酯14、20、23、27的mAb結合在這里,以及在后續步驟中,所有化合物編號都是指所述a和b異構體兩者。典型地,用總體積為250μLPBS的0.6摩爾(用于動力學測量)或10摩爾(用于體內研究)當量的TCO-NHS結構改性1mgCC49(在PBS中的5mg/mL溶液)。用1M碳酸鈉緩沖液將pH調節到9。在黑暗中在攪拌下室溫實施反應30分鐘。隨后,使用AmiconUltra-15離心裝置用PBS徹底洗滌TCO-改性的mAbs。用四嗪滴定(下文描述)確定每個抗體的TCO基團數量。用載體加入的177Lu放射標記四嗪-DOTA28。將用約0.6摩爾當量的TCO改性的mAb(25μg)與3摩爾當量的177Lu-28(0.5nM)反應。將用約10摩爾當量的TCO改性的mAb(25μg)與15摩爾當量的177Lu-28(2.5nM)反應。在50μLPBSpH7.4中在37℃實施反應10分鐘。通過SDS-PAGE和磷光體成像儀(phosphorimager)分析反應混合物,并從對應所述mAb的譜帶中的放射性確定反應產率。計數用AIDAImageAnalyzer軟件(Raytest)定量。發現TCO-mAb結合產率為80-90%。mAb放射標記向在50μLPBS中的足量[125I]碘化鈉(5-15MBq)加入1μL的1mg/mLBolton-Hunter試劑(N-琥珀酰亞胺基-3-[4-羥基苯基]丙酸酯(SHPP))在DMSO中的溶液和25μL的4mg/mL氯胺-T(N-氯4-甲基苯磺酰胺,鈉鹽)在PBS中的溶液。將所獲得的溶液混合10-20秒,向小瓶中加入5μLDMF和100μL甲苯,125I-SHPP被提取在有機相中,然后將其轉移到玻璃小瓶中。在溫和N2流下將甲苯吹出,之后加入CC49-TCO溶液(0.1-0.5mg在50-250μLPBS中),用1M碳酸鹽緩沖液將pH調節到9,并在溫和振蕩下在RT培育反應混合物30分鐘。培育后,通過radio-ITLC確定標記產率。然后將所述粗反應混合物加載到Zeba離心脫鹽柱上,所述Zeba離心脫鹽柱用鹽水溶液預先平衡(pre-equilibrated)。用20μL鹽水溶液沖洗所述反應小瓶并將所述沖洗液也加載到所述柱上。Zeba純化后,通過放射-ITLC、放射-HPLC和SDS-PAGE確定125I-CC49-TCO溶液的放射化學純度;通過BCA檢測確定蛋白質濃度。典型地,采用這個過程,對于純化的125I-CC49-TCO物種獲得大于70%的125I-SHPPmAb結合和>98%的放射化學純度。反應速率用載體加入的177Lu以3MBq/μg的比活度放射標記四嗪-DOTA28。使177Lu-28(33nM)與用1eq.的TCO改性的增加濃度的CC49(0.33、1和1.67μM)在200μLPBS、pH7.4中在37℃反應5分鐘。在選定的時間(15、30、45、60、90、120、180和300秒)抽取20μL試樣并用四嗪6(圖5;在Rossin等AngewChemIntEd2010,49,3375中描述)(1.5μL,5mg/mL在DMF中)猝滅。通過SDS-PAGE和磷光體成像儀分析等分量的各個混合物,并從對應所述mAb的譜帶中的放射性確定環加成產率。計數使用AIDAImageAnalyzer軟件(Raytest)定量。表1:CC49-TCO結構和177Lu-28之間反應的二級動力學常數。O=醚連接;C=氨基甲酸酯(carbamate)連接實施例6反式-環辛烯體內穩定性用CC49-PEG10-TCO-O主要(CC49-23a;8.4TCO每CC49;300μg/100μL每小鼠)、CC49-PEG10-TCO-C次要(CC49-27b;3.3TCO每CC49,μg/100μL每小鼠)和無間隔體的CC49-TCO-O主要(CC49-20a;8.0TCO每CC49,300μg/100μL每小鼠)對無腫瘤小鼠(n=3每組)進行靜脈注射。在選定的時間點(從注射后1小時到不超過4天)從隱靜脈(venasaphena)抽取血液試樣并收集在包含肝素的小瓶中。稱重所述血液試樣并加入過量的111In-四嗪28。在37℃培育20分鐘后,用PBS將血液等分試樣稀釋10倍并通過SDS-PAGE分析。從對應所述mAb的譜帶中的放射性確定環加成產率,并用AIDAImageAnalyzer軟件定量計數。在三個單獨的小鼠組(n=3)中評估在各個時間點的血液中存在的mAb的量(%ID/g血液),其中所述小鼠被注射相應的125I-CC49-TCO(300μg/100μL每小鼠,約0.2MBq)。所述數據(對于mAb清除進行校正)歸一化到在t=0時的100%IDTCO。該實驗的結果表明,除了由于mAb從血液清除導致的TCO生理減少外,在被注射了其中所述TCO通過PEG10間隔體被結合到CC49的結構的兩組小鼠中,循環反應性TCO基團的量隨著時間存在進一步減少(圖13A和B)。對于被注射了其中所述TCO不經間隔體被連接到所述mAb的結構的小鼠則不是這樣的情況(圖13C)。這表明了在體內無間隔體CC49-TCO結構相對于所述CC49-PEG10-TCO結構的更高穩定性。實施例7生物分布實驗通過用具有和不具有PEG10間隔體的125I-標記的CC49-TCO結構(100μg/100μL每小鼠,約0.2MBq),并在24或72小時后用111In-四嗪28(21μg/75μL每小鼠,約0.8MBq)對攜帶腫瘤的小鼠(n=3)進行靜脈注射實施雙同位素生物分布實驗。給予四嗪三小時后,用異氟醚麻醉所述動物并通過頸脫位處死。通過心臟穿刺抽取血液,并采收、剝離干(blotteddry)并稱重感興趣的器官和組織。在γ-計數器中測量所述試樣連同標準物的放射性以確定所述%ID/克。對于125I和111In分別將所述能量窗設定為10-80keV和100-510keV。放射標記的mAbs和111In-四嗪的分布在表2-5中示出。表2:雙同位素生物分布數據注射177Lu-四嗪28(21μg/75μL每小鼠,約0.5MBq)3小時后,給予125I-CC49-PEG10-TCO-O主要(CC49-23a;100μg/100μL每小鼠,約0.2MBq)27小時或99小時后。數據以%ID/克±SD給出。表3:單一同位素生物分布數據注射111In-四嗪28(21μg/75μL每小鼠,約0.5MBq)3小時后,給予CC49-TCO-O主要(CC49-20a;100μg/100μL每小鼠)27小時或75小時后。數據以%ID/克±SD給出。表4:雙同位素生物分布數據注射111In-四嗪28(21μg/75μL每小鼠,約0.5MBq)3小時后,給予125I-CC49-TCO-C次要(CC49-14b;100μg/100μL每小鼠,約0.2MBq)27小時或75小時后。數據以%ID/克±SD給出。表5:雙同位素生物分布數據注射111In-四嗪28(21μg/75μL每小鼠,約0.5MBq)3小時后,給予125I-CC49-TCO-O次要(CC49-20b;100μg/100μL每小鼠,約0.2MBq)27小時或75小時后。數據以%ID/克±SD給出。所述生物分布數據證實了缺少所述間隔體的CC49-TCO結構(constructs)相對于第一代CC49-PEG10-TCO-O主要23a的更高體內穩定性。在注射所述四嗪前用CC49-PEG10-TCO-O主要(CC49-23a)預處理24小時的小鼠中,由于所述TCO和所述四嗪的之間的反應,所述腫瘤中存在的大量mAb(15.81±0.22%ID/克)導致3.09±0.01%ID/克的177Lu積累(表3)。不幸的是,177Lu-四嗪與血液中循環的大量mAb-TCO的反應(5.55±1.85%ID/克)還導致低的腫瘤對血液比(T/B=3.1±1.0)。當在所述mAb4天后給予所述放射標記的四嗪時,由于mAb的清除,所述T/B比值顯著改善(21.3±4.0)。但是,此時也觀察到約四倍低的177Lu-四嗪在腫瘤中的積累(0.86±0.39%ID/克)。絕對四嗪積累的這種減少并非mAb-TCO從組織釋放的結果(腫瘤中仍存在10.99±4.24%ID/克125I-mAb),而合理地是由注射之間的4天期間TCO的體內降解所導致。相反,當用缺少所述PEG10間隔體的CC49-TCO結構預處理所述小鼠時,觀察到mAb注射3天后111In-四嗪的腫瘤攝取相對于mAb注射1天后沒有減少(表4-6)。同時,由于mAb-TCO從血液清除,所述T/B比從約2增加到超過4。在這段時間內腫瘤結合的TCO的得到保持的反應性表明其相對于所述第一代CC49-PEG10-TCO的更高穩定性。實施例8成像實驗接受100μg不含PEG10間隔體(spacer)的CC49-TCO結構3或4天后用111In-四嗪28(21μg/75μL每小鼠,20-50MBq)注射攜帶腫瘤的小鼠。約1小時后,麻醉所述小鼠并放置在裝備有用于麻醉的鼻錐和用于呼吸監控的傳感器的動物床上。注射四嗪2小時后采用四頭多針孔小動物SPECT/CT成像體系(NanoSPECT,BioscanInc.)實施單光子發射計算機斷層成像(Singlephotonemissioncomputedtomography,SPECT)。使用1.4mm直徑的針孔和120-140秒的采集時間/視圖(24次投影)實施所述SPECT采集(總共1小時)。對于111In將所述能量窗設定在245keV±15%和171keV±20%。第一次掃描兩到四天后,通過過量麻醉對所述小鼠進行安樂死并過夜獲取第二次SPECT/CT掃描。每次SPECT期之前實施CT掃描(2秒/投影,360次投影)以獲得關于放射性分布的解剖學信息。所述采集之后,使用制造商的軟件(InVivoScope1.39,patch1)迭代重建數據。對于腫瘤、肝臟、腎臟和大腿肌肉,手工繪制感興趣的區域(ROIs),一式三份。用填充了已知量的111In的體模來標定用于組織放射性定量的掃描儀。圖13A-C中分別示出了注射了CC49-TCO-O次要(CC49-20b)、CC49-TCO-O主要(CC49-20a)和CC49-TCO-C次要(CC49-14b)的三只小鼠在代表性的時間尺度上的圖像。圖13:(左)給予(A)CC49-TCO-O次要(CC49-20b),(B)CC49-TCO-O主要(CC49-20a)或(C)CC49-TCO-C次要(CC49-14b)(100μg/100μL每小鼠)3-4天后,注射111In-四嗪28(21μg/75μL每小鼠,約40MBq)2小時后活小鼠的SPECT/CT投影。(右),所述第一次成像期2-4天后相同小鼠的死后SPECT/CT掃描。白色箭頭表示腫瘤。小鼠中縱向SPECT/CT研究顯示用CC49-TCO-O(主要和次要兩種形式)預處理3天后腫瘤中的高111In-四嗪攝取,并因此證實這些TCO-結構的體內穩定性(圖13A和B)。此時(左圖),大多數非腫瘤結合的mAb-TCO已經從循環清除,并因此由于111In-四嗪的尿排泄除了所述腫瘤以外唯一可見的器官是腎臟和膀胱。重要的是,在血液和富血液的器官例如肝臟中觀察不到放射性。非常低的背景放射性是相對于使用第一代CC49-PEG10-TCO(見R.Rossin,P.RenartVerkerk,SandraM.vandenBosch,R.C.M.Vulders,l.Verel,J.Lub,M.S.Robillard,AngewChemIntEd2010,49,3375)獲得的結果的根本改進,并將轉化為對病人中骨髓和其它劑量限制器官的更低劑量。值得注意的是,所述用CC49-TCO-O預處理的小鼠的腫瘤在注射111In-四嗪72-96小時后仍是高放射性的,表明所述TCO-四嗪環加成產物的高的體內穩定性。在晚的時間點,在所述小鼠腎臟內仍可見一些活性,并且一些在肝臟中積累,合理地是由于抗原脫落和由于注射四嗪時仍在血液內循環的所述mAb的清除所導致。當目標是癌癥病人中的預靶向的RIT時,腫瘤中放射性保持延長的時間也是非常重要的。實際上,治療性放射性核素被結合到靶組織的時間越長,遞送到腫瘤的劑量越高。出人意料地,在給予CC49-TCO-C次要后注射了111In-四嗪的小鼠中觀察到相當低的放射性攝取(uptake),可能是兩次處理之間的4天延遲所導致(圖13C)。但是,也是在這種情況中,在早期掃描2天之后,腫瘤中的信號仍存在,證實由所述體內Diels-Alder反應獲得的環加成產物的體內穩定性。實施例9該實施例涉及在反式環辛烯的不同位置上的各種取代基的電腦模擬(insilico)測試。對于不同取代的環辛烯,使用MOPAC軟件(CambridgestwareMopacProversion8.03)計算了HOMO能量。結果在以下表6中提供。表6通過Mopac對不同取代基確定的HOMO能量。在這里(a)代表直立;(e)代表平伏。碳原子根據以下結構進行編號:實施例10四嗪28的替代性合成過程,和相應的Gd-絡合物,28-Gd的合成。參考圖15,其示出了合成路線。5-氧代-5-(6-(6-(吡啶-2-基)-1,4-二氫-1,2,4,5-四嗪-3-基)吡啶-3-基氨基)戊酸(30)。根據文獻過程(M.L.Blackman,M.Royzen,M.J.Fox,J.Am.Chem.Soc.2008,130,13518-13519)合成了6-(6-(吡啶-2-基)-1,4-二氫-1,2,4,5-四嗪-3-基)-吡啶-3-胺(29)。將29(428mg,1.69mmol)和戊二酸酐(231mg,2.03mmol)在THF(10mL)中的混合物在氬氣惰性氣氛下在60℃加熱40小時。冷卻后,用THF(5mL)洗滌橙色沉淀并干燥以獲得橙色固體形式的30(537mg,87%)。5-氧代-5-(6-(6-(吡啶-2-基)-1,2,4,5-四嗪-3-基)吡啶-3-基氨基)戊酸(6)將化合物30(166mg;0.452mmol)懸浮在醋酸(3mL)中并加入亞硝酸鈉(93.5mg;1.36mmol)。觀察到快速著色成紫色懸浮液。攪拌15分鐘后,過濾所述反應混合物,用水(2×6mL)和丙酮(3mL)洗滌,并干燥,以獲得紫色固體形式的產物(152mg;92%)。(37,41-二氧代-41-((6-(6-(吡啶-2-基)-1,2,4,5-四嗪-3-基)吡啶-3-基)氨基)-3,6,9,12,15,18,21,24,27,30,33-十一氧雜-36-氮雜四十一烷基)氨基甲酸叔丁酯(31)。將PyBOP(148mg,0.284mmol)加入在0℃的6(94.4mg,0.258mmol)、氨基-PEG10-氨基-Boc(150mg,0.233mmol)和N,N-二異丙基乙基胺(新蒸餾,100mg,0.774mmol)在DMF(2mL)中的攪拌混合物中。使所述混合物加溫至室溫并繼續攪拌15分鐘。蒸發所述透明的、深紅色溶液至干燥,并將產物重新溶解在氯仿(5mL)中,用0.2MKH2PO4(pH=4.5,3×3mL)和飽和Na2CO3(2×3mL)洗滌,然后在二乙基醚(20mL)中沉淀。通過離心收集并通過在二氧化硅上使用甲醇在氯仿中的梯度(0-10%)的柱色譜純化所述沉淀,提供紫色蠟狀固體形式的31(148mg,64%)。2,2’,2’’-(10-(2,40,44-三氧代-44-((6-(6-(吡啶-2-基)-1,2,4,5-四嗪-3-基)吡啶-3-基)氨基)-6,9,12,15,18,21,24,27,30,33,36-十一氧雜-3,39-二氮雜四十四烷基)-1,4,7,10-四氮雜環十二烷-1,4,7-三基)三乙酸(28)。將產物31(72.3mg,0.0727mmol)溶解在DCM(1mL)中,并加入TFA(1mL)。在室溫攪拌所述混合物1小時。蒸發后,將剩余物溶解在乙腈(1.5mL)中并在二乙基醚(15mL)中沉淀。通過過濾分離紫色沉淀從而以定量的產率提供32的TFA-鹽。將其溶解在DMF(1.5mL)中,并加入DOTA-NHS(69.6mg,0.075mmol)和N,N-二異丙基乙基胺(新蒸餾,44mg,0.341mmol)。在室溫攪拌所述混合物30分鐘。蒸發透明的、深紅/紫色溶液后,將粗材料溶解在水中并通過制備HPLC純化。冷凍干燥后獲得28(80.5mg,87%產率)。28的GdIII-絡合物,(28-GdIII)將化合物28(204mg,0.160mmol)溶解在醋酸銨水溶液(0.1M,pH=5.5,5mL)中,并加入醋酸釓(III)水合物(96.8mg,0.239mmol)。在室溫攪拌所述混合物30分鐘,接著立即通過制備HPLC純化。冷凍干燥后,獲得紫色固體形式的GdIII-絡合物(188mg,82%產率)。MS(ESI,m/z):計算值C57H89N13O20Gd+([M+H]+):1433.56,實測值:1433.58。實施例11新模型四嗪探針的化學合成合成了包含可選擇的四嗪部分的幾種四嗪模型探針。參考圖16,其示出了所述合成路線。3-(5-丁酰氨基-2-吡啶基)-6-(5-(三氟甲基)-2-吡啶基)-1,2,4,5-四嗪(35)在氬氣惰性氣氛下將2-氰基-5-三氟甲基-吡啶(200mg,1.16mmol)、2-氰基-5-氨基-吡啶(300mg,2.52mmol)和硫磺(80mg,2.52mmol)在乙醇(2mL)中攪拌。加入水合肼(0.60g;12.0mmol)并將所述混合物在80℃加熱過夜。使反應混合物冷卻并加入水(2mL)。離心獲得固體,用水/乙醇=1/2洗滌所述固體并將其干燥以提供135mg粗產物33。向合并的離心上清液加入水,導致進一步批次的粗材料(188mg)的沉淀,通過離心將其分離并干燥。將該粗胺產物(33)和丁酸酐(285mg;1.80mmol)在THF(5mL)中攪拌并在65℃加熱過夜。濃縮所述反應混合物,并在己烷/二乙基醚=3/1中攪拌剩余物。用玻璃過濾器過濾該懸浮液并通過使用己烷/丙酮混合物作為洗脫液的二氧化硅柱色譜純化剩余物,獲得90mg粗產物34(約60%純度)。將未精制的酰胺化的二氫四嗪34(62mg)懸浮在THF(1.5mL)和水(2.0mL)的混合物中。攪拌的同時,加入NaNO2(88mg;1.28mmol),并然后在0℃滴加硫酸(130mg;1.33mmol)在水(1mL)中的溶液。觀察到快速著色成紅色懸浮液。攪拌3分鐘后,用氯仿和水稀釋所述反應混合物,留下紫色沉淀,所述紫色沉淀通過用玻璃過濾器過濾分離。濃縮濾出液中的有機層并用二乙基醚稀釋以誘導紫色固體第二次收獲的沉淀。用氯仿和二乙基醚的混合物磨碎(triturated)合并的固體,并通過過濾分離以提供純四嗪35(約25mg,8%總產率,從2-氰基-5-三氟甲基-吡啶計算)。1HNMR(CDCl3/CD3OD):δ=9.2(s,1H),8.9(d,1H),8.75(多重信號,3H),8.3(d,1H),2.45(t,2H),1.8(m,2H),1.05(t,3H)ppm.19FNMR(CDCl3/CD3OD):δ=-62.9ppm.LC-MS/PDA:色譜中的一個峰,m/z=390.2(M+H+)和800.8(2M+Na+),λmax=329和526nm。3-(5-氟-2-吡啶基)-6-(5-丁酰胺基-2-吡啶基)-1,2,4,5-四嗪(38)在氬氣惰性氣氛下在乙醇(1.5mL)中攪拌2-氰基-5-氟-吡啶(100mg,0.82mmol)、2-氰基-5-氨基-吡啶(200mg,1.68mmol)和硫磺(55mg,1.72mmol)。加入水合肼(0.35g;7.0mmol)并在90℃加熱所述混合物過夜。使所述反應混合物冷卻并加入乙醇(5mL)。用玻璃過濾器過濾獲得固體,用己烷洗滌并然后干燥,以提供90mg粗產物36。接著,在THF(1.5mL)中攪拌該粗胺產物36和丁酸酐(93mg;0.59mmol)并在65℃加熱。在氬氣下反應過夜后,將所述反應混合物冷卻,用一些己烷(約3mL)稀釋并用玻璃過濾器(glassfilter)過濾。通過應用己烷/丙酮混合物作為洗脫劑的二氧化硅柱色譜純化剩余物,獲得純的酰胺化的產物37(約28mg,10%總產率,從2-氰基-5-氟-吡啶計算)。1HNMR(CDCl3/CD3OD):δ=9.25(bs,1H,NH),8.7(s,1H),8.5(s,1H,NH),8.4(多重信號,2H),8.2(m,1H),8.1(m,1H),7.95(m,1H),7.5(m,1H),2.4(t,2H),1.75(m,2H),1.05(t,3H)ppm.13CNMR(CDCl3/CD3OD):δ=172.8,161.6,159.0,146.6,146.1,143.5(d),141.6,139.3,136.8,136.6,136.4,127.0,124.0,123.8,122.6,121.5,38.9,18.8,13.5ppm(由于C-F耦合,一些碳的信號是雙重峰)。LC-MS/PDA:色譜中的一個峰,m/z=342.1(M+H+),λmax=288nm。將所述酰胺化的二氫四嗪37(28mg;0.082mmol)懸浮在THF(2mL)和水(2mL)的混合物中。攪拌的同時,在0℃滴加NaNO2(85mg;1.23mmol),和硫酸(120mg;1.23mmol)在水(2mL)中的溶液。觀察到快速著色成紫色懸浮液。攪拌3分鐘后,加入氯仿和水。用水洗滌所述紫色氯仿層兩次,然后濃縮。在少量氯仿中攪拌固體剩余物,然后向其中加入己烷。將幾乎無色的上清液倒出,干燥固體以獲得21mg紫色粉末38(產率:75%)。1HNMR(CDCl3/CD3OD):δ=8.8(多重信號,5H),7.75(m,1H),2.45(t,2H),1.8(m,2H),1.05(t,3H)ppm.13CNMR(CDCl3/CD3OD):δ=173.4,162.9,162.6,162.4,159.8,146.0,143.2,141.3,139.7和139.4,138.8,126.9,125.8(d),125.2,124.4,124.2,39.0,18.7,13.5ppm(由于C-F耦合,一些碳的信號是雙重峰)。19FNMR(CDCl3/CD3OD):δ=-120.4ppm.LC-MS/PDA:色譜中的一個峰,m/z=340.2(M+H+),λmax=324和529nm。3-(2-吡啶基)-6-甲基-1,2,4,5-四嗪(40)在氬氣惰性氣氛下在乙醇(5mL)中攪拌2-氰基吡啶(500mg,4.8mmol)、乙脒鹽酸鹽(2.00g,21.2mmol)和硫磺(155mg,4.8mmol)。加入水合肼(2.76g;55.2mmol)并在20℃攪拌所述混合物過夜。過濾渾濁的混合物并蒸發濾出液至干燥,獲得2.9g橙色的粗產物39。接著,將該粗產物(800mg)懸浮在THF(3mL)和醋酸(4mL)的混合物中。在0℃加入NaNO2(2.0g;29.0mmol)在水(3mL)中的溶液。觀察到立刻著色成紅色/紫色懸浮液。在0℃攪拌5分鐘后,加入氯仿和水。用水洗滌所述紫色氯仿層兩次,然后濃縮。在氯仿和己烷的1:1混合物中攪拌固體剩余物,然后過濾。濃縮濾出液并通過應用氯仿/丙酮混合物作為洗脫液的二氧化硅柱色譜純化所述粗產物,獲得純產物40(48mg,21%總產率,從2-氰基吡啶計算)。1HNMR(CDCl3):δ=8.96(d,2H),8.65(d,2H),7.99(t,2H),7.56(dd,3H),3.17(s,3H)ppm.13CNMR(CDCl3):δ=168.1.163.6,150.9,150.3,137.4,126.3,123.9,21.4ppm.LC-MS/PDA:色譜中的一個峰,m/z=174.3(M+H+),λmax=274和524nm。3,6-雙(4-吡啶基)-1,2,4,5-四嗪(42)在氬氣惰性氣氛下在90℃加熱4-氰基吡啶(858mg;8.24mmol)和一水合肼(1.24g;24.7mmol)16小時。使所述混合物冷卻到室溫并然后用水(3mL)稀釋。過濾橙色沉淀(41)并用水(3mL)洗滌,隨后將其溶解在DMSO(10mL)中。向該溶液加入DDQ(372mg;1.64mmol)。觀察到立即著色成深紅色溶液。60分鐘后,加入飽和碳酸氫鈉溶液(20mL)并用氯仿(3次30mL)提取產物。用Na2SO4干燥合并的有機層并將其蒸發至干燥以獲得粉色固體形式的標題化合物(52mg;5%總產率,從4-氰基吡啶計算)。1H-NMR(CDCl3):δ8.97(d,4H),8.52(d,4H)ppm.LC-MS/PDA:色譜中的一個峰,m/z=237.2(M+H+),λmax=271和523nm。實施例12額外的反式-環辛烯結構的化學合成參考圖17,其詳細描述合成路線。(E-主要)-2,5-二氧代吡咯烷-1-基2-(環辛-4-烯-1-基氧基)醋酸酯(44a)向冰冷卻的10a(1.73g,13.73mmol,包含約10%順式異構體)在THF(40mL)中的溶液加入在油中的氫化鈉(60%,2.60g,65.0mmol)。在RT攪拌所述混合物15分鐘,然后將其加熱到50℃達1小時。在冰中冷卻所述混合物,加入溴乙酸(2.50g,17.9mmol)。在冰中攪拌所述懸浮液1小時,然后在約25℃攪拌64小時,加入更多的THF以保持可攪拌的懸浮液(總體積約100mL)。在50℃加熱1小時后,冷卻所述混合物,緩慢加入水。通過旋轉蒸發除去大部分THF并加入更多的水(總體積50mL)。用MTBE(2×75mL)提取含水混合物,并用25mL水洗滌有機層。用16g檸檬酸酸化合并的水層,并用2×75mLMTBE提取產物。干燥并旋轉蒸發留下剩余物,通過柱色譜(40gSiO2)將其純化。合并產物級分,從含有痕量MTBE的庚烷重結晶剩余物。這提供了810mg的產物43a(810mg,4.40mmol,32%,含有少量順式異構體)。1H-NMR(CDCl3):δ1.4–2.45(m,10H),3.1–3.2(m,1H),3.9–4.1(AB,2H),5.3–5.65(m,2H)。將產物43a溶解在30mL二氯甲烷中。加入N-羥基琥珀酰亞胺(715mg,6.22mmol)并在冰中冷卻混合物。加入DCC(1.42g,6.89mmol)并在冰中攪拌所述混合物30分鐘,然后在RT攪拌4小時。過濾、旋轉蒸發和進行在40g硅膠上使用庚烷/EtOAc梯度的色譜提供44a。合并產物級分,并從含有少量MTBE的庚烷重結晶。刮擦之后產物結晶。過濾后獲得180mg的產物(0.64mmol,15%,含有約20%順式異構體)。(E-次要)-2,5-二氧代吡咯烷-1-基2-(環辛-4-烯-1-基氧基)醋酸酯(44b)向冰冷卻的10b(0.78g,6.19mmol)在THF(30mL)中的溶液加入在油中的氫化鈉(60%,0.94g,23.5mmol)。在RT攪拌所述混合物15分鐘,然后加熱到50℃達1小時。在冰中冷卻所述混合物,加入溴乙酸(1.41g,10.14mmol)。在約25℃攪拌懸浮液20小時(試樣表明還未發生耦合),然后在55℃攪拌6小時,在25℃攪拌3天,并在55℃再攪拌6小時。通過旋轉蒸發除去大部分THF,并加入50mLMTBE,接著加入冰和25mL水。分離層,并用30mLMTBE提取水層。用25mL水洗滌相繼的有機層。在冰中冷卻合并的水層,加入50mLMTBE,接著加入5.1g檸檬酸。分離層,并用50mLMTBE提取水層。干燥和旋轉蒸發留下剩余物(43b),將其就這樣用于下一步驟。1H-NMR(CDCl3):δ1.2–2.45(m,10H),3.65–3.75(m,1H),4.1(s,2H),5.45–5.65(m,2H)。將產物43b溶解在30mL二氯甲烷中。加入N-羥基琥珀酰亞胺(1.60g,13.91mmol)并在冰中冷卻所述混合物。加入DCC(3.11g,15.10mmol)并在冰中攪拌混合物30分鐘,然后在RT攪拌3小時。過濾、旋轉蒸發和進行在40g硅膠上使用甲苯然后是二氯甲烷作為洗脫液的色譜提供了44b,將其與溴乙酸的NHS酯混合。將混合物溶解在25mLMTBE中,向所述溶液加入25mL庚烷。攪拌2小時后,過濾混合物(固體是溴乙酸的NHS酯)。旋轉蒸發濾出液,將剩余物溶解在溫MTBE中,加入一些庚烷,然后將溶液冷卻到RT。其沉淀出產物。過濾提供30mg產物44b(0.11mmol,2%)。(E-主要)-2,5-二氧代吡咯烷-1-基2-(環辛-4-烯-1-基氧基)-2-苯基乙酸酯(46a)向10a(3.0g,23.8mmol,含有<10%順式異構體)在THF(40mL)中的溶液加入在油中的60%NaH(3.0g,75.0mmol)。在RT攪拌混合物10分鐘,然后將其加熱到50℃達1.5小時。在冰中冷卻后,分份加入DL-2-溴苯乙酸(3.87g,18.0mmol)。向稠糊狀物加入THF(20mL)并在25℃攪拌懸浮液18小時。在55℃通過旋轉蒸發除去大部分THF并加入50mLMTBE。在冷水中冷卻混合物并加入一些冰,接著加入50mL水。分離層并用50mLMTBE提取水層。用25mL水洗滌相繼的有機層。在冰中冷卻合并的水層,加入50mLMTBE,接著加入10g檸檬酸。分離層并用50mLMTBE提取水層。干燥和旋轉蒸發留下5.05g產物45a,將其就這樣用于下一步驟。將產物45a溶解在50mL二氯甲烷中。加入N-羥基琥珀酰亞胺(2.51g,21.8mmol)并在冰中冷卻所述混合物。加入DCC(5.03g,24.4mmol)并在冰中攪拌混合物15分鐘,然后在RT攪拌3小時。過濾、旋轉蒸發和進行在55g硅膠上使用甲苯/二氯甲烷梯度的色譜提供2.8g46a(7.83mmol,33%基于10a,包含約5%順式異構體)。(E-次要)-2,5-二氧代吡咯烷-1-基2-(環辛-4-烯-1-基氧基)-2-苯基乙酸酯(46b)向在0℃的10b(1.0g,7.9mmol)在THF(60mL)中的溶液加入在油中的60%NaH(1.26g,31.5mmol),并將混合物加熱到50℃1.5小時。冷卻到0℃后,將DL-2-溴苯乙酸(2.22g,10.3mmol)以在THF(5mL)中的溶液形式加入,并在RT劇烈攪拌粘性懸浮液16小時。再加熱到40℃24小時后,將混合物倒入檸檬酸(9.2g)在水(100mL)中的溶液中。用MTBE(3×50mL)提取含水混合物,用Na2SO4干燥并蒸發溶劑以獲得黃色油。通過柱色譜(SiO2,CH2Cl2/MeOH2%)純化提供產物、未反應的TCO和副產物的混合物。通過溶解在MTBE和水(其用33%NaOH溶液堿化)中實現了進一步純化。水層用MTBE洗滌,用檸檬酸酸化并然后用MTBE(3×)提取。用Na2SO4干燥合并的有機層,溶劑蒸發后獲得黃色油形式的化合物45b(463mg,1.8mmol,23%產率)。向在0℃的45b(463mg,1.8mmol)和N-羥基琥珀酰亞胺(250mg,2.2mmol)在THF(9mL)中的溶液加入DCC(372mg,1.8mmol)在THF(2mL)中的溶液。在RT攪拌所述反應混合物16小時,之后通過過濾除去沉淀。在真空中濃縮濾出液并通過柱色譜(SiO2、庚烷/EtOAc梯度25%-40%)純化以提供無色漿形式的46b(451mg,1.3mmol,71%產率)。(E-主要)-2,5-二氧代吡咯烷-1-基2-(環辛-4-烯-1-基氧基)-2-甲基丙酸酯(49a)向在冰浴中冷卻的10a(3.0g,23.8mmol)在THF(120mL)中的溶液加入在油中的60%氫化鈉(3.8g,95mmol)。除去冰浴并在RT攪拌所述混合物30分鐘,然后在50℃攪拌1小時。在冰中冷卻后,緩慢加入DL-2-溴丙酸(3.3mL,35.6mmol)。在所述加入期間混合物變得十分粘稠,用THF(50mL)將其稀釋。所述添加完成后,除去冰浴并在RT攪拌所述反應混合物16小時。通過NMR跟蹤反應的進度,其在16小時后呈現40%的轉化率。然后在35℃將混合物再攪拌24小時,獲得84%的轉化率。在真空中濃縮反應混合物,用MTBE稀釋并然后用水(200mL)猝滅(quenched)。分離層并用檸檬酸(22.5g)在水(60mL)中的溶液酸化水層。用MTBE(3×200mL)提取所述水層。用Na2SO4干燥合并的有機層并蒸發所述溶劑以提供包含47a的混合物(5.19g)。所述材料未經進一步純化用于接下來的步驟。向在-70℃的二異丙胺(13mL,92mmol)在THF(200mL)中的溶液緩慢加入在己烷中的2.5M正丁基鋰(32mL,80mmol)。然后將所述混合物緩慢加溫到-20℃并再次冷卻到-70℃。以在THF中的溶液形式加入化合物47a(5.19g)并使所述混合物加溫到-20℃。在該溫度加入碘甲烷(10.7mL,172mmol)并將所述混合物加溫到5℃。取試樣并根據NMR分析所述反應完成了。將反應混合物倒入檸檬酸(40g)在水(200mL)中的溶液并用MTBE(3×150mL)提取。用檸檬酸水溶液和鹽水洗滌合并的有機層。用Na2SO4干燥并蒸發所述溶劑后獲得黃色油(9.4g)。通過柱色譜(SiO2,CH2Cl2/MeOH梯度1%-4%)純化提供黃色漿形式的純48a(1.15g,5.4mmol,兩個步驟23%產率)。1H-NMR(CDCl3):δ1.1–2.45(m)和1.4(2s)(16H),3.2–3.3(m,1H),5.3–5.7(m,2H)。向在0℃的48a(1.15g,5.4mmol)和N-羥基琥珀酰亞胺(622mg,5.4mmol)在THF(27mL)中的溶液加入DCC(1.12g,5.4mmol)在THF(5mL)中的溶液。加入之后,將所述混合物加溫至RT并在此溫度攪拌16小時。用MTBE稀釋混合物,并過濾掉沉淀。蒸發溶劑后,通過柱色譜(SiO2,庚烷/EtOAc梯度20%-40%)純化剩余物。從庚烷/EtOAc結晶提供無色晶體形式的49a(707mg,2.3mmol,42%產率)。(E-次要)-2,5-二氧代吡咯烷-1-基2-(環辛-4-烯-1-基氧基)-2-甲基丙酸酯(49b)向在冰浴中冷卻的10b(1.5g,11.9mmol)在THF(60mL)中的溶液加入在油中的60%氫化鈉(1.9g,48mmol)。除去冰浴并在50℃攪拌所述混合物1.5小時。在冰中冷卻后,緩慢加入DL-2-溴丙酸(1.7mL,17.8mmol)。添加期間混合物變得非常粘稠并用THF(35mL)稀釋。添加完成后除去冰浴并在RT攪拌所述反應混合物16小時。然后將所述混合物加熱到43℃保持24小時。在真空中濃縮所述反應混合物,用MTBE稀釋并然后用水(200mL)猝滅。分離層,并用檸檬酸在水中的溶液酸化水層。用MTBE(3×100mL)提取所述水層。用Na2SO4干燥合并的有機層并蒸發溶劑以提供包含47b的混合物(2.26g)。所述材料未經進一步純化用于接下來的步驟。向在-70℃的二異丙胺(5.6mL,39.6mmol)在THF(100mL)中的溶液緩慢加入在己烷中的2.5M正丁基鋰(13.5mL,33.8mmol)。然后緩慢將所述混合物加溫到0℃并再次冷卻到-70℃。以在THF中溶液的形式加入化合物47b(2.26g)并將所述混合物加溫到-20℃。在該溫度下加入碘甲烷(4.6mL,73.9mmol)并使所述混合物加溫到15℃。取樣并根據NMR分析所述反應完成了。將反應混合物倒入檸檬酸(20g)在水(140mL)中的溶液中并用MTBE(3×100mL)提取。用檸檬酸水溶液和鹽水洗滌合并的有機層。用Na2SO4干燥并蒸發溶劑后獲得黃色漿(4.6g)。通過柱色譜(SiO2,CH2Cl2/MeOH梯度1%-4%)重復純化提供白色結晶固體形式的純48b(181mg,0.85mmol,2個步驟7%產率)。向在0℃的48b(181mg,0.85mmol)和N-羥基琥珀酰亞胺(98mg,0.85mmol)在THF(5mL)中的溶液加入DCC(175mg,0.85mmol)在THF中的溶液。加入后,將所述混合物加溫到RT并在該溫度攪拌5小時。用MTBE稀釋混合物并過濾掉沉淀。蒸發所述溶劑后,通過柱色譜(SiO2,庚烷/EtOAc梯度25%)純化剩余物以提供白色固體形式的49b(257mg,0.83mmol,98%產率)。(E-主要)-2,5-二氧代吡咯烷-1-基4-(環辛-4-烯-1-基氧基)苯甲酸酯(52a)向冰冷卻的10a(3.45g,27.38mmol)在THF(50mL)的溶液加入在油中的氫化鈉(60%,2.5g,62.5mmol)。在冰中將所述混合物攪拌15分鐘,然后加熱到50℃達1小時。在冰中冷卻所述混合物,并用5分鐘的時間加入溶解在5mLTHF中的4-氟苯甲酰氯(1.72g,10.84mmol)。在25℃攪拌所述混合物2天(試樣的NMR表明存在很多產物,但也存在4-氟苯甲酸的TCO-酯),然后在50℃攪拌3小時。用水冷卻所述混合物,并緩慢加入10mL水,接著加入2g氫氧化鈉和5mL水。通過旋轉蒸發除去大部分溶劑,加入THF和2g氫氧化鈉并將所述混合物在50℃加溫4小時。通過旋轉蒸發除去大部分溶劑并向剩余的糊狀物加入甲醇。將所述混合物在50℃加溫2小時,接著在55℃旋轉蒸發。用50mL水稀釋余下的懸浮液。加入MTBE(100mL),分離層并用25mL水洗滌有機層。所述有機層包含a.o.反式環辛烯醇。用20g檸檬酸處理合并的冰冷卻的水層,并用2×75mLMTBE提取產物。干燥和旋轉蒸發留下固體剩余物,其由產物和4-氟苯甲酸的混合物組成。將所述固體用40mL甲醇加溫。將水(15-20mL)緩慢加入所述溫溶液中直到其變得渾濁。過濾并冷卻濾出液沉淀出產物。過濾、用1/1甲醇/水洗滌和在真空下干燥提供0.827g想要的產物51a(3.36mmol,基于4-氟苯甲酰氯為30%)。用DCC(1.46g,7.08mmol)處理冰冷卻的主要反式酸51a(1.14g,4.63mmol)和N-羥基琥珀酰亞胺(725mg,6.30mmol)在50mL二氯甲烷中的混合物。在冰中攪拌所述混合物1小時,然后在RT攪拌3小時。過濾、旋轉蒸發和實施在40g硅膠上使用庚烷/EtOAc梯度的色譜提供52a級分(其被順式異構體污染)。合并接下來的產物級分,并用庚烷和醋酸乙酯的混合物將其加溫到60℃。使懸浮液冷卻到RT,然后過濾。這提供了0.973g產物52a(2.83mmol,61%)。(E-次要)-2,5-二氧代吡咯烷-1-基4-(環辛-4-烯-1-基氧基)苯甲酸酯(52b)向冰冷卻的10b(2.82g,22.38mmol)在THF(40mL)中的溶液加入在油中的氫化鈉(60%,2.0g,50mmol)。在冰中攪拌所述混合物15分鐘,在RT攪拌30分鐘,然后加熱到50℃達1小時。在冰中冷卻所述混合物,并用15分鐘的時間加入溶解在8mLTHF中的4-氟苯甲酰氯(1.72g,10.84mmol)。在RT攪拌所述混合物18小時(試樣的NMR表明存在許多反式環辛烯醇),然后在50℃攪拌6小時,在25℃攪拌3天,和在50℃再攪拌1小時。緩慢加入2.0g氫氧化鈉在5mL水中的溶液,接著加入5mL水。通過旋轉蒸發除去大部分THF并向所獲得的糊狀物加入40mL甲醇。將混合物在50℃加溫3小時,加入20mLTHF并繼續加熱4小時。在25℃攪拌所述混合物過夜(NMR表明仍存在少量的酯),然后在50℃加熱4小時,接著旋轉蒸發。用50mL水稀釋余下的懸浮液。加入MTBE(100mL),分離層并用25mL水洗滌有機層。所述有機層包含a.o.反式環辛烯醇。用15g檸檬酸處理合并的冰冷卻的水層并用2×75mLMTBE提取產物。干燥和旋轉蒸發留下固體剩余物,其由產物和4-氟苯甲酸的混合物組成。用40mL甲醇加溫所述固體。向所述溫溶液緩慢加入水(25mL),接著使其冷卻到RT并然后攪拌過夜。過濾,用1/1甲醇/水洗滌并在真空下干燥提供0.76g想要的產物51b(3.09mmol,基于4-氟苯甲酰氯為28%)。用DCC(0.95g,4.61mmol)處理冰冷卻的所述次要反式酸51b(0.66g,2.68mmol)和N-羥基琥珀酰亞胺(460mg,4.0mmol)在50mL二氯甲烷中的混合物。在冰中攪拌所述混合物1小時,然后在RT攪拌16小時。過濾、旋轉蒸發和實施在30g硅膠上使用二氯甲烷作為洗脫液的色譜。將產物溶解在5mL醋酸乙酯中。加入庚烷并在60℃部分旋轉蒸發所述溶液直到出現沉淀。加入庚烷并在60℃攪拌所述混合物5分鐘,然后使其冷卻到RT。過濾提供0.437g產物52b(1.27mmol,47%)。(E-主要,次要)-2,5-二氧代吡咯烷-1-基2-(1-甲基環辛-4-烯-1-基)醋酸酯(58)將35%的過氧化氫溶液(25mL,300mmol)加入醋酸鈀(45%Engelhard,1.0g,2mmol)、苯醌(432mg,4mmol)和1,5-環辛二烯(27mL,200mmol)的混合物中。在30℃攪拌所述混合物5天直到NMR分析顯示所述起始材料95%的轉化率。將所述混合物倒入Et2O(1L)中并加入水(1L)。用33%的NaOH溶液緩慢堿化所述混合物同時用冰冷卻。分離層并用Et2O(2×)提取水層。用1NNaOH洗滌合并的有機層兩次并用Na2SO4干燥。小心蒸發溶劑提供黃色油形式的粗53(16.1g,130mmol,65%產率)。將1.6M正丁基鋰在己烷中的溶液(45mL,72mmol)加入二異丙胺(14mL,100mmol)在THF(250mL)中的溶液中,冷卻到-80℃。將所述混合物逐漸加溫到0℃并然后冷卻到-80℃。加入膦酰基乙酸三乙酯(triethylphosphonoacetate)(15mL,75mmol)在THF(100mL)中的溶液,并在-70℃攪拌所述混合物45分鐘。然后加入53(6.21g,50mmol)在THF(50mL)中的溶液并將混合物緩慢加溫到RT。16小時后將所述混合物進一步加熱至回流8小時直到NMR分析顯示完全轉化。將所述混合物倒入水(250mL)中并用MTBE(3×)提取。用鹽水洗滌合并的有機層并用Na2SO4干燥。旋轉蒸發溶劑并通過柱色譜(SiO2,庚烷/EtOAc5%)純化后獲得無色油形式的54(4.74g,24mmol,49%產率)。將1.6M的甲基鋰溶液(59mL,94mmol)加入在冰浴中冷卻的碘化銅(I)(9.53g,50mmol)在Et2O(21mL)中的懸浮液中。在0℃在真空中濃縮灰色溶液并用CH2Cl2剝離(stripped)兩次。將剩余物懸浮在冷的CH2Cl2(100mL)中并在緩慢加入TMSCl(4.0mL,46.5mmol)前冷卻到-80℃。然后通過滴加的方式加入54(4.74g,24mmol)在CH2Cl2(60mL)中的溶液,并用2小時使所述混合物加溫到RT。在將所述混合物猝滅入(quenchedinto)飽和的NH4Cl水溶液(150mL)中之前將其在RT另外攪拌16小時。在RT攪拌混合物并加入氨(50mL)。過濾混合物,用CH2Cl2(3×75mL)提取濾出液。用水洗滌并用Na2SO4干燥合并的有機層。旋轉蒸發溶劑并通過柱色譜(SiO2,庚烷/EtOAc3%)純化提供無色油形式的55(4.47g,21mmol,89%產率)。1H-NMR(CDCl3):δ0.8–1.9(m),1.05(s)和1.25(t)(16H),2.15–2.3(m,2H),4.0–4.2(q,2H),5.4–5.55(m,1H),5.65–5.75(m,1H)。用庚烷/Et2O3:1v/v(約500mL)的混合物填充燒瓶,所述燒瓶裝配有汞燈并且與泵和填充有10%硝酸銀(I)浸漬的硅膠(36g,在底部具有一些普通等級硅膠)的柱連接。加入55(4.47g,21.3mmol)和苯甲酸甲酯(2.7mL,21.3mmol)在少量Et2O中的溶液后,開啟汞燈。使被照射的溶液連續流動經過所述柱照射20小時后,在反應器內容物的NMR分析中沒有觀察到起始材料,用在庚烷中的30%MTBE洗滌所述柱。用氨處理柱內容物并用CH2Cl2(3×)提取。用Na2SO4干燥合并的有機層,在蒸發溶劑并通過柱色譜(SiO2,庚烷/EtOAc梯度3%>4%)純化后獲得無色油形式的化合物56(1.41g,6.7mmol,31%產率)。所述主要和次要異構體無法分離,因此所述異構體以混合物形式被進一步使用。1H-NMR(CDCl3):δ0.8–1.9(m),1.0(s)和1.25(t)(16H),2.1–2.4(m,2H),4.0–4.2(q,2H),5.5–5.65(m,2H)。將氫氧化鋰一水合物(84mg,2.0mmol)在水(1mL)和EtOH(1mL)中的溶液加入56(210mg,1.0mmol)中。為了更好地溶解所述化合物,加入THF(1mL)和MeOH(2mL)。在RT攪拌16小時后轉化仍未完全。加入額外的氫氧化鋰(85mg)并將所述混合物加熱到45℃4小時,在30℃16小時和在50℃4小時直到轉化完全。在真空中濃縮所述混合物并用檸檬酸溶液中和。用MTBE(3×)提取并用Na2SO4干燥后,旋轉蒸發溶劑提供黃色油形式的57(150mg,0.82mmol,82%產率)。1H-NMR(CDCl3):δ1.0(2s,3H),1.3–2.0(m,10H),2.1–2.45(m,2H),5.5–5.65(m,2H)。向冰冷卻的57(150mg,0.82mmol)和N-羥基琥珀酰亞胺(113mg,0.98mmol)在THF(5mL)中的溶液加入DCC(170mg,0.82mmol)在THF(1mL)中的溶液。在RT攪拌所述混合物16小時并然后用MTBE稀釋。通過過濾除去沉淀并在蒸發溶劑并通過柱色譜(SiO2,庚烷/EtOAc梯度10%>30%)純化后獲得無色油形式的化合物58(197mg,0.71mmol,86%產率)。1H-NMR(CDCl3):δ1.05和1.1(2s,3H),1.35–2.35(m,10H),2.4(s,1H),2.5-2.75(AB,1H),2.85(s,4H),5.5–5.65(m,2H)。實施例13新TCOs的反應動力學關于其對四嗪28的反應性評估了所述新合成的TCO結構。實施例5中描述了過程。表7:CC49-TCO結構和177Lu-28之間的反應的二級動力學常數。對于所有TCOs,在所述主要(平伏)和所述次要(直立)異構體之間都存在深遠和一致的反應性差異,后者比前者具有大得多的反應性。此外,TCO58的高反應性支持了沒有至抗體的連接體的直立位置中的取代基仍提供所述反式環辛烯環的增加的反應性。實施例14新模型四嗪探針的穩定性和反應性關于它們在水中的反應性和穩定性評價了包含可選擇的四嗪部分的幾種四嗪模型探針。四嗪類的水解穩定性測試用PBS緩沖液(3mL)稀釋10μL的特定四嗪在DMSO中的溶液(25mM),并過濾溶液。使用UV光譜監測在525nm處的吸收帶的降低,并從該數據測定水解速率和半衰期。四嗪類對反式-環辛-4-烯-1-醇(次要異構體,10b)的反應性實施競爭實驗以確定特定四嗪和3-(5-乙酰胺基-2-吡啶基)-6-(2-吡啶基)-1,2,4,5-四嗪(將其選擇作為參比四嗪)在與反式-環辛-4-烯-1-醇(次要異構體,10b)的逆電子需求Diels-Alder反應中的反應性比。向乙腈(0.100mL)加入5μL所述特定四嗪在DMSO(25mM)中的溶液和5μL所述參比四嗪在DMSO(25mM)中的溶液。用水(0.9mL)稀釋該混合物,通過LC-MS/PDA分析測定兩種四嗪的絕對量。接著,加入反式-環辛-4-烯-1-醇(次要異構體,10b)在DMSO(25μL2.5mM)中的溶液,并攪拌所述混合物5分鐘。再次通過LC-MS/PDA分析測定兩種四嗪的絕對量,并計算兩種四嗪的轉化率。從這些轉化率確定兩種四嗪的反應性比。表8:模型四嗪探針的反應性和穩定性從表8清楚地看到在四嗪上的取代基對穩定性和反應性都有深遠的影響,因此能夠用于為特定應用特制的探針的設計。實施例15主要vs次要TCO,20avs20b與四嗪探針28的體外反應性差異實施該實驗以證明在兩種試劑的低濃度下更高TCO反應性轉化為與放射標記的四嗪的更高反應產率。為了這一目的,我們使用了連接到CC49的TCO主要20a(k2=19600±1400M-1s-1)和TCO次要20b(k2=136700±2300M-1s-1)(對于主要和次要分別為6.1和6.6TCOs/分子)。在PBS中稀釋兩種mAb溶液以獲得1×10-5M到1×10-8M的TCO濃度,并使其與載體加入的177Lu-四嗪(相對于mAb為1當量)在37℃反應1分鐘。等分量的反應混合物(20μL)然后用過量四嗪6猝滅,加入非還原性的試樣緩沖液并通過SDS-PAGE分析。從對應所述mAb的譜帶中的放射性測定環加成產率并用AIDAImageAnalyzer軟件定量計數。重復所述實驗三次。如對雙分子反應所預期的那樣,培育1分鐘后溶液中所述兩種反應性物種的濃度的降低轉化為更低的反應產率(圖18)。但是,當使用所述直立TCO20b時,在微摩爾濃度和更低濃度下,由于與所述四嗪的更快的反應動力學,反應產率明顯高于使用平伏TCO20a獲得的那些。實施例16TCO20b的體內穩定性實施該實驗以擴展實施例6中描述的系列體內TCO穩定性測量。在那個系列中,發現用PEG間隔體結合到CC49的TCOs主要和次要(23a和27b)在體內不穩定,同時發現不含PEG間隔體的TCO主要(20a)在體內穩定至少4天(圖13)。在所述新實驗中,遵循相同的過程,我們測試了不含PEG間隔體的CC49-結合的TCO次要(20b,6.9當量結合)的體內穩定性。圖19中示出了對mAb清除進行校正的所述體內穩定性數據。發現所述CC49-結合的TCO20b在體內僅緩慢降解,進一步支持了具有至抗體的短連接體的TCOs在體內比通過更長連接體結合的TCOs更穩定的發現。實施例17用不同量TCO20b部分官能化的CC49抗體的血液動力學和生物分布實施了一系列研究以評估每個CC49分子上TCO(20b)基團數量對血液循環和腫瘤靶向的影響。預期用過多的TCO部分改性抗體會導致血液循環半衰期的下降并將影響CC49-TCO結構的腫瘤攝取。CC49-TCO結構的血液動力學(見圖20和21)用攜帶0、4.9、9.3或15.8個TCO(20b)基團/mAb的125I-CC49對雌性裸Balb/C小鼠(20-25g體重,CharlesRiverLaboratories,n=3)進行注射(100μg/100μL每小鼠,約0.5MBq)。在選擇的時間點(5分鐘,3、6小時,1、2、3天)從隱靜脈抽取血液試樣,稱重并用1mLPBS稀釋。注射mAb四天后,麻醉所述動物并通過頸脫位處死,通過心臟穿刺收集血液并采收、剝離干、稱重感興趣的器官和組織,并加入1mLPBS。在γ-計數器(gamma-counter,WizardII,PerkinElmer)中連同標準物測量所有試樣中的放射性以確定每克組織的百分比注射劑量(%ID/g)。CC49-TCO結構在血液中的半衰期用GraphPadPrism(version5.01)計算(通過將血液曲線擬合成two-phasedecay)。表9:CC49-TCO(20b)的動力學血液參數。CC49-TCO(20b)結構的腫瘤靶向用采用0、8.5、12.7或18.7個TCO20b基團/mAb官能化的125I-CC49對攜帶腫瘤的小鼠(見方法部分;100mm3腫瘤尺寸)進行注射(100μg/100μL每小鼠,0.2-0.4MBq,n=4)。注射mAb4天后處死接受了125I-CC49的小鼠。注射mAb72小時后用111In-DOTA-四嗪28對接受了125I-CC49-TCO20b的小鼠進行注射(相對于mAb為25當量,約0.8MBq)并在注射四嗪后3個小時將其處死。處死時,通過心臟穿刺收集血液并采收、剝離干、稱重感興趣的器官和組織,并加入1mLPBS。在γ-計數器(Wizard3,PerkinElmer)中采用雙同位素方案(對于125I和111In能量窗分別設定為10-80keV和100-510keV)連同標準物測定所有試樣中的放射性以確定每克組織的百分比注射劑量(%ID/g)。表10:注射mAb3天后(*除外)125I-CC49-TCO20b結構在攜帶腫瘤的小鼠中的生物分布。數據以平均%ID/g±SD(n=4)給出*mAb注射4天后處死小鼠**低免疫反應性。表11:111In-四嗪28(注射3小時后)在用CC49-TCO20b結構預處理的小鼠中的生物分布。數據以平均%ID/g±SD(n=4)給出從表9-11中的數據可以清楚看到TCO改性比不應超過9。實施例18CC49-TCO44b的血液動力學和體內穩定性血液動力學用125I-標記的CC49對無腫瘤的小鼠(n=3/組)進行注射(100μg每小鼠,0.2-0.4MBq),CC49已用7.5TCO44b基團改性。在選擇的時間點(1、3、6、24、48和72小時)從隱靜脈抽取血液試樣并收集在含有肝素的小瓶中。注射mAb四天后,麻醉所述小鼠并通過頸脫位處死。通過心臟穿刺抽取血液并采收、剝離干、稱重感興趣的器官和組織,加入1mLPBS,在γ-計數器(Wizard3,PerkinElmer)中連同標準物進行計數以確定每個器官的百分比注射劑量(%ID)。在該研究中,TCO-改性的CC49呈現出比未改性的CC49稍快的清除(圖22)。對于CC49-TCO和CC49,從曲線下的面積計算的血液中的半衰期(T1/2=ln2×AUC/C0)分別為22.2小時和26.3小時。我們將這歸因于所述mAb上Lys殘基的官能化。作為較短血液循環的結果,在所述實驗結束時,在大多數器官中還觀察到較少量的125I-CC49-TCO,所述器官在計數之前不灌注(perfused)(圖23)。體內穩定性用125I-標記的CC49-TCO44b(220μg每小鼠,0.4MBq)對單獨組的無腫瘤小鼠(n=3)進行注射。在選定的時間點(1、3、6、24、48和72小時)從隱靜脈抽取血液試樣(約50微升)并收集在包含肝素的小瓶中。注射mAb四天后,麻醉所述小鼠并通過頸脫位處死。通過心臟穿刺抽取血液并除去胃和甲狀腺,剝離干并在γ-計數器中連同標準物一起計數以確定每個器官的百分比注射劑量(%ID)。這些器官中的低125I攝取(在胃中為0.17±0.03%ID和在甲狀腺中為0.88±0.33%ID)證實所述放射標記的mAb在4天評價期間保持了體內標記物。稱重所述血液試樣,用PBS稀釋至100μL并加入過量的以0.1MBq/μg比活性放射標記的載體加入的177Lu-四嗪28。在37℃培育所述混合物20分鐘并以400×g離心5分鐘以分離血細胞。然后向ZebaDesalt離心柱(0.5ml,40kDaMW截止,Pierce)施加30μL上清液。離心后,從筒洗脫高MWDiels-Alder反應產物,同時保留過量的四嗪。在γ-計數器中采用雙同位素方案(對于125I為10-80keV窗和對于177Lu為155-380keV窗)測量所述洗出液中包含的放射性。將僅包含177Lu-四嗪的血清試樣用于校正來自所述筒的177Lu泄露。關于放射性衰減校正125I計數并然后計算177Lu/125I比。177Lu/125I比的減小表明小鼠血液試樣中存在的過量177Lu-四嗪和125I-CC49-TCO之間更低的反應產率,并且因此表明所述TCO基團的體內失活。圖24作為%完好TCO示出了177Lu/125I比隨著時間的變化(t=0時歸一化為100%)。值得注意的是,mAb注射后不超過48小時所述TCO44b基團看起來在體內是完全穩定的(97.8±1.6%完好TCO),而在更晚的時間點觀察到一些降解(注射mAb4天后為80.4±1.8%完好TCO)。實施例19177Lu-四嗪28在用CC49-TCO44b預靶向的攜帶腫瘤的小鼠中的生物分布用125I-CC49對攜帶腫瘤的小鼠(見方法部分;100mm3腫瘤尺寸;n=4)進行注射(100μg每小鼠,約0.2MBq),所述125I-CC49用7.5TCO44b基團官能化。注射mAb三十和48小時后,小鼠接受一個劑量的清除劑(半乳糖-MSA-四嗪,160μg/劑量)接著2小時后使其接受177Lu-四嗪28(相對于所述mAb為10當量,約0.5MBq)。注射四嗪三小時后,麻醉所述小鼠并通過頸脫位處死,通過心臟穿刺抽取血液,采收感興趣的器官和組織并剝離干。稱重所有收集的試樣并加入1mLPBS。在γ-計數器中采用雙同位素方案(對于125I為10-80keV窗口和對于177Lu為155-380keV窗口)連同標準物測定試樣放射性以確定每克組織的百分比注射劑量(%ID/g)。生物分布數據顯示125I-CC49-TCO44b的高腫瘤攝取。所述腫瘤攝取高于用其它TCO結構(見下文)獲得的那些,合理地是因為用7.5TCO-44b基團官能化的CC49的長血液循環。相反,由于給予了兩劑量的清除劑,所述清除劑捕捉循環CC49-TCO并將其導向肝臟,在那里其被迅速代謝,因此在所有其它器官中mAb保留都是低的。作為腫瘤中較高mAb攝取的結果,177Lu-四嗪的攝取也明顯高于在先前實驗中獲得的那些(見下文)。而且,由于在給予四嗪之前的追蹤步驟(chasestep)中除去了非腫瘤結合的CC49-TCO,在所有其它器官中的177Lu攝取是可忽略的。作為四嗪尿排泄的結果只有腎臟呈現相當高的177Lu保留。這導致除腎臟外在所有考慮的器官中高的靶對非靶比。值得注意的是,腫瘤和血液中存在的34±4%和10±1%的TCOs已經分別與四嗪反應。表12:注射177Lu-四嗪28(8.5μg/80μL每小鼠,約0.5MBq)3小時后,給予125I-CC49-TCO-44b(100μg/100μL每小鼠,約0.2MBq)50小時后的雙同位素生物分布數據。數據以%ID/克±SD或腫瘤/器官比±SD(n=4)給出。
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1