干擾素高分子結合體IFN-PMPC的制備及其應用的制作方法
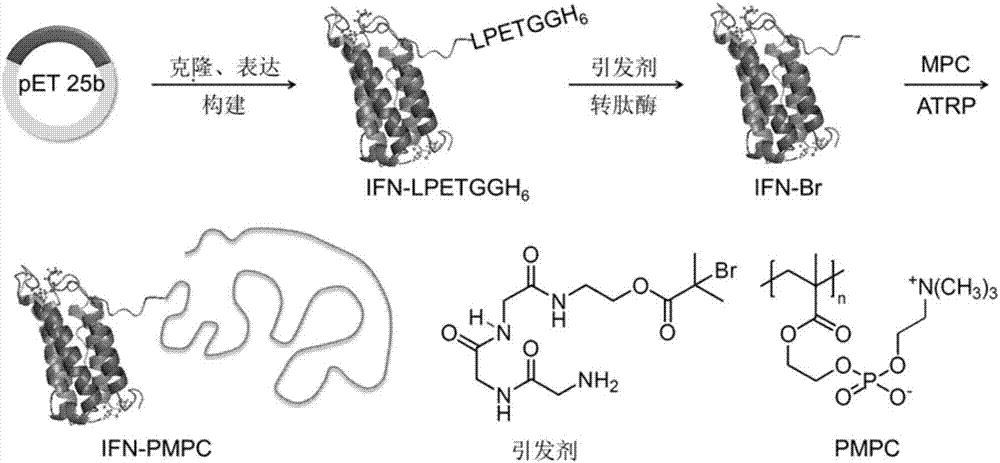
本發明屬于生物醫藥領域,具體涉及制備干擾素高分子結合體ifn-pmpc的制備及其應用。
背景技術:
:蛋白質已經廣泛應用于生物醫藥顯影、靶向治療以及臨床診斷等多個領域。單獨使用蛋白質存在半衰期短、穩定性差等問題。將蛋白質與高分子相連制備蛋白質-高分子結合體,可以有效的提高蛋白質的溶解性、穩定性、藥代動力學和治療功效并降低其免疫原性。傳統的蛋白質-高分子結合體的合成方法是將預先制備好的高分子與蛋白質相連,往往存在著偶聯位點不確定、效率低、產率差、產物難以分離、質量控制差、活性難以保持等諸多難題。干擾素-α2(ifn-α2)是病毒復制和腫瘤細胞生長的強效抑制劑,已被成功用于治療病毒性肝炎和癌癥等疾病。但是,ifn經系統注射給藥后循環半衰期非常短,需要頻繁給藥且在高濃度下才能達到預期的療效,從而導致一些毒副作用,同時給患者帶來沉重的經濟負擔。用聚乙二醇(peg)修飾ifn,是改善其藥代動力學和提高其療效的有效措施,被稱為長效干擾素。然而,目前的peg化干擾素存在如反應產率低、結合位點和偶聯化學計量難以控制、生物活性嚴重降低等弊端。另外,peg存在免疫原性,多次給藥后,會產生peg抗體,加速其在體內的清除,改變藥物在體內的分布和代謝。因此,研發反應條件溫和、步驟簡單的、沒有免疫原性的位點特異性修飾方法對干擾素及其他藥用蛋白質尤為重要。技術實現要素:本發明的一個目的是提供一種制備蛋白質-高分子結合體的方法。本發明提供的方法,為如下1)或2):1)所示的方法包括如下步驟:將蛋白質-引發劑結合體、高分子單體、cucl、cucl2、1,1,4,7,10,10-六甲基三乙烯四胺在緩沖液中進行聚合反應,得到蛋白質-高分子結合體;所述蛋白質-引發劑結合體為引發劑和蛋白共價連接得到的產物;所示的方法中聚合反應所用的緩沖液為ph值為7.4、濃度為10mm的pbs水溶液,具體配方為:2.684gna2hpo4·12h2o、0.34gnah2po4·2h2o、8.19gnacl溶解于1l水中得到的溶液。2)所示的方法為在引發劑的作用下將高分子化合物和蛋白通過寡聚甘氨酸連接,得到蛋白質-高分子結合體;所述高分子化合物為通過高分子單體聚合得到。上述兩種方法中,所述蛋白為干擾素,所述干擾素選自干擾素α或其融合蛋白、干擾素β或其融合蛋白、干擾素γ或其融合蛋白、干擾素λ或其融合蛋白;或所述蛋白具體為序列表中序列2所示的干擾素α融合蛋白。所述高分子單體為2-甲基丙烯酰氧乙基磷酸膽堿;所述高分子化合物為聚2-甲基丙烯酰氧乙基磷酸膽堿。所述引發劑為原子轉移自由基聚合反應的引發劑、可逆加成一斷裂鏈轉移聚合的引發劑、開環異位聚合的引發劑或開環加成聚合的引發劑;所述原子轉移自由基聚合反應的引發劑的結構式如下式1:其中,r為h或(ch2)nch3,n為≥1的整數;x為cl或br或i;m為≥1的整數;n為≥1的整數;所述原子轉移自由基聚合引發劑具體為2-溴-2-甲基丙酸2-(2-(2-(2-氨基乙酰胺基)乙酰胺基)乙酰胺基)乙酯鹽酸鹽;所述引發劑共價連接到所述蛋白的c端或n端;1)所示的方法中,所述引發劑共價連接到所述蛋白的c端或n端;或所述引發劑具體共價連接到所述蛋白的c端;上述方法中,1)所示的方法中,所述蛋白質-引發劑結合體、所述2-甲基丙烯酰氧乙基磷酸膽堿、所述cucl、所述cucl2和所述1,1,4,7,10,10-六甲基三乙烯四胺的摩爾比為1:200-10000:10-500:10-2000:10-4000;所述蛋白質-引發劑結合體、所述2-甲基丙烯酰氧乙基磷酸膽堿、所述cucl、所述cucl2和所述1,1,4,7,10,10-六甲基三乙烯四胺的摩爾比具體為1:1000-4000:10-500:10-2000:10-4000;所述蛋白質-引發劑結合體中所述蛋白和所述引發劑的偶聯比為1:1。上述方法中,所述蛋白質-引發劑結合體、所述2-甲基丙烯酰氧乙基磷酸膽堿、所述cucl、所述cucl2和所述1,1,4,7,10,10-六甲基三乙烯四胺的摩爾比為1:2000:25:75:125。1)所示的方法中,上述蛋白質-引發劑結合體為引發劑在蛋白的c端,二者 通過形成酰胺鍵而形成結合體,具體按照包括如下步驟的方法制備:將蛋白(ifn-lpetggh6蛋白)、轉肽酶(sortasea-h6蛋白)、2-(2-(2-(2-氨基乙酰胺基)乙酰胺基)乙酰胺基)乙基2-溴-2-甲基丙酸酯、cacl2在ph7.4濃度為50mmtris·hcl水溶液中混合,反應,得到干擾素-引發劑結合體ifn-br;上述ifn-lpetggh6蛋白、sortasea-h6蛋白、2-(2-(2-(2-氨基乙酰胺基)乙酰胺基)乙酰胺基)乙基2-溴-2-甲基丙酸酯、cacl2的摩爾比為2:1:50:200。上述2)所示的方法,包括如下步驟:將所述引發劑和所述高分子單體發生聚合反應,得到連接寡聚甘氨酸的高分子聚合物;再將所述連接寡聚甘氨酸的高分子聚合物與所述蛋白共價連接,得到蛋白質-高分子結合體。上述2)所示方法中,所述聚合反應為將引發劑、2-甲基丙烯酰氧乙基磷酸膽堿、cucl、cucl2和1,1,4,7,10,10-六甲基三乙烯四胺在緩沖液中進行聚合反應,得到連接寡聚甘氨酸的高分子聚合物;所述聚合反應采用的緩沖液為ph值為7.4、濃度為10mm的pbs水溶液;所述共價連接為將所述蛋白質、轉肽酶、所述連接寡聚甘氨酸的高分子聚合物、和cacl2在緩沖液中進行反應,得到蛋白質-高分子結合體;所述共價連接的反應中的緩沖液為ph7.4濃度為50mmtris·hcl水溶液。上述2)所示方法中,所述聚合反應中,所述引發劑、所述2-甲基丙烯酰氧乙基磷酸膽堿、所述cucl、所述cucl2和所述1,1,4,7,10,10-六甲基三乙烯四胺的摩爾比為1:200-10000:10-500:10-2000:10-4000;或所述引發劑、所述2-甲基丙烯酰氧乙基磷酸膽堿、所述cucl、所述cucl2和所述1,1,4,7,10,10-六甲基三乙烯四胺的摩爾比具體為1:2000:25:75:125;所述共價連接中,所述蛋白質(ifn-lpetggh6蛋白)、所述轉肽酶(sortasea-h6蛋白)、所述連接寡聚甘氨酸的高分子聚合物(連接三甘氨酸的聚2-甲基丙烯酰氧乙基磷酸膽堿)和cacl2的摩爾比為2:1:40:200。所述緩沖液為ph7.4濃度為50mmtris·hcl水溶液;上述方法中,所述聚合反應在低氧或者惰性氣體氛圍下進行;所述聚合反應的時間是5分鐘至24小時,所述聚合反應的溫度是0-80℃。由上述方法制備的蛋白質-高分子結合體也是本發明保護的范圍。上述的蛋白質-高分子結合體在制備抗腫瘤或抗病毒產品中的應用也是本發明保護的范圍。本發明另一個目的是提供一種抗腫瘤或抗病毒產品。本發明提供的產品,其包括上述的蛋白質-高分子結合體。本發明通過利用生物化學和高分子化學技術,在遠離干擾素的c-末端進行修飾并 通過優化過的原子轉移自由基聚合(atrp)技術原位高效生長出pmpc(聚2-甲基丙烯酰氧乙基磷酸膽堿),產生位點特異和單修飾的干擾素-高分子結合體(即ifn-pmpc)。這種原位atrp技術反應條件溫和、產率高、純化步驟簡單、成本低,能夠有效保持生物活性并提高體外穩定性,為蛋白質修飾提供一種通用的方法。附圖說明圖1為干擾素-高分子結合體ifn-pmpc合成路線的示意圖。圖2為通過鎳柱親和層析獲得ifn-lpetggh6。圖3為通過鎳柱親和層析獲得sortasea-h6。圖4為原位atrp引發劑aebm的合成及純化過程。圖5為ifn-br合成及純化過程分析。圖6為maldi-tof分析ifn-lpetggh6和ifn-br的分子量。圖7為lc-ms/esi分析了ifn-br的特異性修飾情況。圖8為ifn-pmpc合成及純化過程。圖9為gpc分析ifn-pmpc。圖10為顯示了通過1hnmr分析ifn-pmpc結合體。圖11為分析原位atrp合成ifn-pmpc的對照試驗。圖12為通過“graftingto”技術獲得ifn-pmpc圖13為sds-page分析蛋白酶k處理ifn-pmpc。圖14為dls分析ifn-pmpc和ifn-α的水合半徑。圖15為cd分析ifn-pmpc和ifn-α的二級結構。圖16為mtt法測定ifn-pmpc、派羅欣和ifn-α的體外生物活性。圖17為利用das軟件中房室消除二房模型分析ifn-pmpc、派羅欣和ifn-α的藥物代謝動力學情況。圖18為ifn-pmpc、派羅欣和ifn-α在腫瘤和其他組織中的分布情況。圖19為ifn-pmpc、派羅欣、ifn-α和生理鹽水抑制腫瘤生長情況。圖20為裸鼠注射ifn-pmpc、派羅欣、ifn-α和生理鹽水后的生存曲線。圖21為裸鼠注射ifn-pmpc、派羅欣、ifn-α和生理鹽水后的體重變化情況。圖22為裸鼠治療結束后腫瘤、心臟、肝臟和腎臟he染色情況。圖23為裸鼠注射ifn-pmpc、派羅欣、ifn-α和生理鹽水后乳酸脫氫酶、肌酸激酶同工酶、谷丙轉氨酶、谷草轉氨酶、肌酐和尿素氮等生理指標變化情況。圖24為裸鼠ifn-pmpc、派羅欣、ifn-α和生理鹽水后紅細胞、白細胞、血紅蛋白和血小板等生理指標變化情況。具體實施方式下述實施例中所使用的實驗方法如無特殊說明,均為常規方法。下述實施例中所用的材料、試劑等,如無特殊說明,均可從商業途徑得到。下述實施例中的質粒pet-25b(+)為生工生物工程(上海)股份有限公司產品。下述實施例中的tb培養基按照如下方法配置:向900ml水中加入蛋白胨12g、酵母提取物24g和甘油4ml,充分溶解后121℃高壓滅菌15min,滅菌后的混合液冷卻至60℃,然后加入100ml滅菌的含170mmol/lkh2po4和0.72mol/lk2hpo4的水溶液。下述實施例中的人burkitt’sb淋巴瘤細胞和人卵巢癌細胞(ovcar-3)均購自中國科學院腫瘤細胞庫。下述實施例中的rmpi-1640培養基為gibco公司產品。下述實施例中的雌性無胸腺(nude)裸鼠為北京維通利華實驗動物技術有限公司產品。雌性無胸腺(nude)裸鼠在下文中簡稱裸鼠。下述實施例中的流程如圖1所示。下述實施例中,利用基質輔助激光解吸電離飛行時間質譜(maldi-tof)、酶標儀、動態光散射(dls)、圓二色譜(cd)等分析手段表征ifn-pmpc和ifn的分子量、相變溫度、水合半徑和二級結構等物理化學性能。選用人burkitt’sb淋巴瘤細胞,測試ifn-pmpc和ifn的體外生物活性,即其體外抗腫瘤細胞增殖的能力;使用裸鼠模型,測試ifn-pmpc和ifn在體內的藥物代謝動力學,利用das3.0藥物代謝動力學分析軟件計算出藥物代謝動力學參數;建立裸鼠腫瘤模型,測試ifn-pmpc和ifn的抗腫瘤效果和藥物分布。下述實施例中的定量實驗,如無特殊說明均設置三次重復,結果取平均值。實施例1、制備干擾素高分子結合體ifn-pmpc的方法一、制備干擾素-引發劑結合體1、制備ifn-lpetggh6融合蛋白和轉肽酶sortasea-h61)重組菌的制備ifn-lpetggh6融合蛋白的氨基酸序列為序列表中序列2,其編碼基因的核苷酸序列為序列表中序列1;轉肽酶sortasea-h6的氨基酸序列為序列表中序列4,其編碼基因的核苷酸序列為序列表中序列3;表達ifn-lpetggh6融合蛋白的重組載體為將序列1所示的ifn-lpetggh6融合蛋白編碼基因插入pet-25b(+)載體的ndei和ecori酶切位點中得到的載體,命名為pet-25b-ifn-lpetggh6;表達sortasea-h6的重組載體為將序列3所示的sortasea-h6蛋白編碼基因插入pet-25b(+)載體的ndei和ecori酶切位點中得到的載體,命名為pet-25b-sortase a-h6。表達ifn-lpetggh6融合蛋白的重組菌為將表達ifn-lpetggh6融合蛋白的重組載體導入e.colibl21rosetta-gami(de3)plyss(invitrogen)中得到重組菌,命名為bl21/pet-25b-ifn-lpetggh6;表達sortasea-h6的重組菌為將表達sortasea-h6的重組載體導入e.colibl21(de3)plyss(novagen)中得到重組菌,命名為bl21/pet-25b-sortasea-h6。2)ifn-lpetggh6蛋白的表達和純化將重組菌bl21/pet-25b-ifn-lpetggh6在50mltb培養基中(含100μg/ml氨芐青霉素),37℃、250rpm條件下震蕩培養過夜。第二天再轉接入1l新鮮tb培養基當中(盛在2l的搖瓶中,氨芐青霉素濃度為100μg/ml)進行大規模培養并誘導表達。具體步驟如下:首先在37℃,200rpm條件下震蕩培養5h,隨后將培養溫度設為18℃,加入異丙基-β-d-硫代半乳糖苷(iptg),終濃度為0.4mm,培養16h,收集培養液。以3000×g離心力離心收集菌體,去除上層培養液,得到重組菌bl21/pet-25b-ifn-lpetggh6菌體。用30ml冰冷pbs重懸bl21/pet-25b-ifn-lpetggh6菌體,再用超聲儀在4℃條件下破碎細胞,然后將大腸桿菌破碎產物在4℃、14000×g離心力下離心15分鐘。在收集的上清液中加入2ml聚乙烯亞胺(pei,10%),再次離心15分鐘。得到的上清液經過0.45μm濾膜過濾后,在akta蛋白純化系統(aktapurifier10,ge)上經過鎳親和層析柱(histraphp5ml)提純,平衡緩沖液為10mmpbs、500mmnacl,5%甘油,洗脫緩沖液為平衡緩沖液加500mm咪唑得到的混合液。收集洗脫峰對應的洗脫液,且用十二烷基硫酸鈉-聚丙烯酰胺凝膠電泳(sds-page)檢測,留約21.1kda的目標產物對應的洗脫液,為ifn-lpetggh6蛋白。sds-page分析樣品由含有5%β-巰基乙醇的laemmli樣品緩沖液配制,濃度為1mg/ml,95℃下加熱5min后,10μl樣品裝載到預制的10%sds-page凝膠中,垂直電泳80~100v電壓下運行90min(電泳液為:25mmtris、250mmglycine、0.1%sds)。凝膠用考馬斯藍g-250染色處理后觀察條帶位置。洗脫所得到的ifn-lpetggh6蛋白經脫鹽柱(hiprep26/10desalting)除去咪唑,同時置換到50mmtris·hcl緩沖液中。純化樣品用sds-page測試純度,并用分光光度計法(nanodrop2000)測定了蛋白的濃度。結果如圖2所示,a為通鎳親和層析純化,uv監測線性洗脫得到目標產物ifn-lpetggh6;b為sds-page分析ifn-lpetggh6純化情況;顯示了ifn-lpetggh6蛋白表達與鎳親和層析柱純化情況。從左至右依次為蛋白標樣、上樣前細胞裂解液、鎳柱流穿液、50mm咪唑洗柱和500mm咪唑洗脫目的蛋白,可以看出,通過 大腸桿菌進行表達、鎳親和層析柱純化獲得純度>95%的ifn-lpetggh6蛋白,產率達到200mg/l培養菌液,可以看出,得到約21.1kda的目標產物。3)sortasea-h6蛋白的表達和純化采用同樣的方法處理重組菌bl21/pet-25b-sortasea-h6,收集洗脫峰對應的洗脫液,且用sds-page檢測。結果如圖3所示,a為通鎳親和層析純化,uv監測線性洗脫得到目標產物sortasea-h6;b為sds-page分析sortasea-h6純化情況;顯示了sortasea-h6蛋白表達與鎳親和層析柱純化情況。從左至右依次為蛋白標樣、上樣前細胞裂解液、鎳柱流穿液、50mm咪唑洗柱和500mm咪唑洗脫目的蛋白,可以看出留約22.9kda的目標產物。2、合成atrp引發劑2-羥基乙基氨基甲酸叔丁醇酯(2.0g,12.5mmol),n,n-二異丙基乙基胺(2.4ml,14mmol,1.1當量)在冰水浴中溶于二氯甲烷(20ml)。在低于0攝氏度條件下滴加2-溴-2-甲基丙酰溴(1.25ml,10mmol),15分鐘滴加完畢。30分鐘后,移除冰水浴,并保持攪拌16小時。旋蒸移除溶劑,產物用硅膠柱純化(二氯甲烷:乙酸乙酯=1:1)。產物為黃色油狀液體,2-溴-2-甲基丙酸2-(2-(叔丁氧羰基)胺基)乙酯(c11h20brno4,2.9g,74.7%).1hnmr(400mhz,cdcl3):δ4.827(s,1h),4.243(m,2h),3.424(m,2h),1.945(s,6h),1.447(s,9h).esi-massm/z:332.1([m+na]+),334.1([m+na]+).2-溴-2-甲基丙酸2-(2-(叔丁氧羰基)胺基)乙酯(2.5g)溶于6m-的氯化氫乙酸乙酯溶液50ml,攪拌2小時。反應結束后,過濾,產物為白色粉末,2-溴-2-甲基丙酸2-氨基乙酯鹽酸鹽(c6h13brclno2,1.95g,98%).esi-massm/z:210.3([m–cl+na]+),212.3([m–cl+na]+).2-(2-(2-叔丁氧羰基)乙酰胺基)乙酰胺基)乙酸(289mg,1mmol),2-溴-2-甲基丙酸2-氨基乙酯鹽酸鹽(246mg,1mmol),edc(288mg,1.5mmol),和n,n-二異丙基乙基胺(175μl,1mmol)溶于氯甲烷(10ml),室溫攪拌16小時。反應結束后,移除溶劑,殘留白色固體一次用4毫升水洗滌2次,1毫升乙醇洗滌3次,然后用4毫升乙酸乙酯洗滌,得到白色粉末狀產物,2-溴-2-甲基丙酸2-(2-(2-(2-叔丁氧羰基)乙酰胺基)乙酰胺基)乙酰胺基)乙酯(c15h26brn3o6,269mg,56%).1hnmr(400mhz,cdcl3):δ4.261(t,2h),3.909(s,2h),3.871(s,2h),3.779(s,2h),3.544(t,2h),1.938(s,6h),1.449(s,9h).esi-massm/z:481.2([m+h]+),483.1([m+h]+),503.2([m+na]+),505.1([m+na]+).2-溴-2-甲基丙酸2-(2-(2-(2-叔丁氧羰基)乙酰胺基)乙酰胺基)乙酰胺基)乙酯(1.08g,2.25mmol)溶于6m的氯化氫乙酸乙酯溶液50ml,攪拌2小時。產 物為白色粉末,2-溴-2-甲基丙酸2-(2-(2-(2-氨基乙酰胺基)乙酰胺基)乙酰胺基)乙酯鹽酸鹽,即atrp引發劑aebm(c12h22brcln4o5,化學結構式如式1所示,0.84g,98%).1hnmr(400mhz,d2o):δ4.196(t,2h),3.938(s,2h),3.815(s,2h),3.794(s,2h),3.456(t,2h),1.813(s,6h).esi-massm/z:381.1([m–cl+h]+),383.0([m–cl+h]+)。式1如下:圖4顯示了原位atrp引發劑aebm的合成及純化過程。最終成功獲得純度>95%的引發劑aebm,產率為40.2%。3、通過轉肽酶a酶催化獲得干擾素-引發劑結合體通過轉肽酶a酶催化在ifn-lpetggh6的c-末端引入atrp引發劑aebm,具體如下:將上述1制備的上述ifn-lpetggh6蛋白、sortasea-h6蛋白、上述2制備的aebm、cacl2在ph7.4濃度為50mmtris·hcl水溶液中混合,反應,得到干擾素-引發劑結合體ifn-α;上述ifn-lpetggh6蛋白、sortasea-h6蛋白、上述2制備的aebm、cacl2的摩爾比為2:1:50:200。具體方法如下:在10ml含有ifn-lpetggh6(200μm)的50mmtris·hcl溶液(溶質為ifn-lpetggh6,溶劑為ph值為7.4、濃度為50mmtris·hcl水溶液)中加入5mmaebm,與10ml含有100μm轉肽酶a和20mmcacl2的tris·hcl溶液混合,室溫反應過夜,得到反應混合物。將反應混合物用陰離子交換層析(hitrapcaptoq5ml)純化,得到ifn-br;純化采用的平衡緩沖液為20mmtris·hcl,ph7.5;洗脫緩沖液為含有1mnacl的20mmtris·hcl水溶液,ph7.5;柱子大小為5ml,流速為2ml/min;收集洗脫峰對應的洗脫液,且用sds-page檢測,留約20.5kda的目標產物對應的洗脫液,為干擾素-引發劑結合體ifn-br,并用pbs經脫鹽柱進一步去除小分子雜質。該反應效率>95%。將干擾素-引發劑結合體ifn-br用sds-page進行了驗證分析,結果如圖5所示,顯示了ifn-br合成及純化過程分析,其中,a:aex分離轉肽酶a催化ifn-lpetggh6與aebm反應的混合物,uv監測線性洗脫得到目標產物ifn-br;b: sds-page分析ifn-lpetggh6與aebm反應前后得到ifn-br的結果。從左到右依次為蛋白標樣、srta-h6、ifn-lpetggh6、反應混合物、洗脫蛋白ifn-br、流穿樣品srta-h6。純化產物ifn-br的分子量用基質輔助激光解吸電離-飛行時間質譜(maldi-tof)測定,所用儀器為4800plusmaldi-tof/toftm分析儀(absciex)。用液相色譜-電噴霧串聯質譜法(lc-ms/esi)分析驗證ifn-br的氨基酸序列及c-末端的特異性修飾,所用儀器為q-exactive液質聯用質譜儀(thermoscientific)。圖6為maldi-tof分析ifn-lpteggh6和ifn-br分子量,分別為21105.8和20531.9,與理論值21105.9和20532.0符合。圖7為ifn-br的c-末端肽段egsggggslpetggg(-br)同位素分布情況,a圖為lc-ms/esi實驗值;b圖為理論預測值,理論值與實驗值吻合。表1顯示了lc-ms/esi分析的ifn-br胰蛋白酶分解后肽段氨基酸序列組成。圖6、圖7和表1表明利用轉肽酶a能夠定點定量獲得干擾素-引發劑結合體ifn-br,引發劑定點修飾在ifn-α的c-末端,干擾素-引發劑結合體ifn-br中蛋白和引發劑的偶聯比為1:1。表1二、干擾素高分子結合體ifn-pmpc的制備與表征1、干擾素高分子結合體ifn-pmpc的制備利用atrp技術在上述干擾素-引發劑結合體ifn-br表面原位合成pmpc,方法包括如下步驟:將干擾素-引發劑結合體ifn-br、mpc、cucl、cucl2、1,1,4,7,10,10-六甲基三乙烯四胺(hmteta)和ph值為7.4、濃度為10mm的pbs水溶液(pbs配方為:2.684gna2hpo4·12h2o、0.34gnah2po4·2h2o、8.19gnacl溶解于1l水中),在密閉條件下反應,得到干擾素高分子結合體ifn-pmpc;干擾素-引發劑結合體ifn-br、mpc、cucl、cucl2、1,1,4,7,10,10-六甲基三乙烯四胺(hmteta)的摩爾比為1:2000:25:75:150;具體如下:在2.5ml含有40μmifn-br的pbs溶液(溶質為ifn-br,溶劑為ph值為7.4、濃度為10mm的pbs溶液)中加入100μmol的mpc,通氮氣15min后,加入預先溶解于1ml雙蒸水中并已除空氣的cucl(2.5μmol),cucl2(7.5μmol)和1,1,4,7,10,10-六甲基三乙烯四胺(hmteta)(12.5μmol),密閉條件下反應1h,然后暴露在空氣中終止反應,得到反應混合物。反應混合物經陰離子交換柱(hitrapcaptoqcolumn,gehealthcare)分離純化出ifn-pmpc,該純化步驟與上述相同:純化采用的平衡緩沖液為20mmtris·hcl,ph7.5;洗脫緩沖液為20mmtris·hcl,1mnacl,ph7.5,柱子大小為5ml,流速為2ml/min;收集洗脫峰對應的洗脫液,且用sds-page檢測,留約60kda的目標產物對應的洗脫液,為ifn-pmpc。計算產率公式為(純化后ifn-pmpc結合體的質量/ifn-lpetggh6的質量)*100%,結果如表2,ifn-pmpc的產率分別為67.0%。表2圖8顯示了ifn-pmpc的合成及純化過程,其中,a為aex分離原位atrp反應的混合物,uv監測線性洗脫得到目標產物ifn-pmpc;b為sds-page分析ifn-br原位atrp生長高分子pmpc,從左往右依次為:標樣、ifn-lpetggh6、ifn-br、反應混合物、純化后的ifn-pmpc。圖9為gpc分析ifn-pmpc結合體。圖10通過1hnmr分析ifn-pmpc結合體。為了驗證pmpc是利用ifn-br上的引發劑在c-末端合成,用不含引發劑的三甘氨酸連接到ifn上得到ifn-α,然后用上述相同的條件進行了對照試驗。圖11顯示了分析atrp合成ifn-pmpc的對照試驗,其中,a:gpc分析ifn-α合成ifn-pmpc的對照試驗。b:sds-page分析,從左至右依次為蛋白標樣、ifn-α進行atrp反應和ifn-α。對照試驗采用與ifn-br相同的反應條件對ifn-α用來進行atrp反應,顯示ifn-α并沒有生長出pmpc,說明該聚合反應僅在連接了atrp引發劑的ifn-br的c-末端進行,而不存在蛋白其他反應基團上的副反應。三、干擾素高分子結合體ifn-pmpc的另一種制備方法為了顯示原位atrp技術的優越性,利用“graftingto”技術合成ifn-pmpc。graftingto技術包含兩步驟:1、合成連接三甘氨酸的聚2-甲基丙烯酰氧乙基磷酸膽堿pmpc按照上述二中的1的制備步驟,將引發劑aebm、mpc、cucl、cucl2、1,1,4,7,10,10-六甲基三乙烯四胺(hmteta)和ph值為7.4、濃度為10mm的pbs水溶液,在密閉條件下反應,得到連接三甘氨酸的pmpc;其中,引發劑aebm、mpc、cucl、cucl2、1,1,4,7,10,10-六甲基三乙烯四胺(hmteta)摩爾比為1:2000:25:75:125;上述反應條件與上述二中的1相同。利用超濾方法除去小分子等雜質,用gpc檢測連接三甘氨酸的pmpc的分子量,約為60kda。2、pmpc與ifn結合得到干擾素高分子結合體ifn-pmpc通過轉肽酶a催化在ifn-lpetggh6的c-末端引入連接三甘氨酸的pmpc,具體 如下:將上述二制備的25μmifn-lpetggh6蛋白、12.5μmsortasea-h6蛋白、500μm上述1制備的連接三甘氨酸的pmpc和10mmcacl2,在ph7.4濃度為50mmtris·hcl水溶液中混合,常溫過夜孵育,得到孵育產物經過純化得到干擾素高分子結合體ifn-pmpc;上述ifn-lpetggh6蛋白、sortasea-h6蛋白、連接三甘氨酸的pmpc、cacl2的摩爾比為2:1:40:200。上述孵育產物經一次陽離子交換柱(hitrapmacrocapspcolumn,gehealthcare,柱子大小為5ml,流速為2ml/min,平衡緩沖液為20mmtris·hcl,ph7.0;洗脫緩沖液為20mmtris·hcl,1mnacl,ph7.0)除去轉肽酶a,洗脫液為陽離子交換產物;將陽離子交換產物經一次陰離子交換柱(hitrapcaptoqcolumn,gehealthcare,柱子大小為5ml,流速為2ml/min,平衡緩沖液為20mmtris·hcl,ph8.0;洗脫緩沖液為20mmtris·hcl,1mnacl,ph8.0)除去未反應的ifn-lpetgh6和pmpc,收集洗脫液為ifn-pmpc;純度>95%,產率為0.9%。圖12為gpc和sds-page分析了“graftingto”技術合成ifn-pmpc,其中,a圖為gpc技術分析“graftingto”技術;b圖為sds-page分析離子交換純化ifn-pmpc,從左至右分別為:蛋白標樣、反應前混合物、反應24h后混合物、第一次陽離子交換柱純化后混合物、第二次陰離子交換柱純化后ifn-pmpc、利用原位atrp技術合成ifn-pmpc。結果表明,利用“graftingto”技術合成ifn-pmpc反應產率低,純化困難,說明原位atrp技術具有產率高、純化簡單、易于放大及應用等顯著優勢。四、干擾素高分子結合體ifn-pmpc的物理化學表征ifn-pmpc(采用上述二制備的結合體ifn-pmpc)的分子量和多分散系數(pdi)用gpc測定,所用儀器為watershplc/gpc系統連接uv檢測器(waters2489)和示差折光檢測器(waters2414)。色譜柱為asahipakgs-520hq和gs-320hq或gs-520hq串聯,流動相為50mmtris·hcl緩沖液(ph7.4),檢測條件為25℃,流速0.5ml/min。不同分子量的窄分布peg標準樣生成的標準曲線用來計算分子量和pdi。為了準確計算高分子pmpc的分子量,先通過蛋白酶降解與原位高分子偶聯的蛋白質再進行表征。具體步驟為:ifn-pmpc結合體(1mg/ml)與蛋白酶k(0.5mg/ml)在50mmtris·hcl,2mmcacl2,ph7.4,45℃孵育12h。圖13顯示了ifn-pmpc可以被蛋白酶k降解ifn,再通過gpc準確表征pmpc分子量,從左往右依次為:蛋白標樣、ifn-pmpc、ifn-pmpc與蛋白酶k反應前、ifn-pmpc與蛋白酶k反應過夜后、蛋白酶k。通過gpc分析,ifn-pmpc的分散系數為1.43,分子量為57kda。在malvernzetasizernano-zs90上用動態光散射(dls)方法測定ifn-pmpc 的水合半徑。樣品稀釋在pbs緩沖液中,測試前經0.22μm孔徑濾膜過濾處理。經dls測試,ifn-lpetggh6、ifn-br和ifn-α的水合半徑為2.4nm,2.3nm和2.3nm,而合成的ifn-pmpc為9.7nm。圖14顯示了dls分析ifn-pmpc。用核磁共振儀(1hnmr)表征蛋白質-高分子的化學結構。ifn-pmpc樣品經冷凍干燥后,溶解于d2o,在jeolecx-400400mhz核磁共振光譜儀上進行分析。通過1hnmr譜圖證實了蛋白分子上成功合成了pmpc。圖10顯示了通過1hnmr分析ifn-pmpc結合體。ifn-pmpc的二級結構用圓二色性譜分析測得。樣品用水溶液稀釋至0.10mg/ml(1.0μm),用pistarπ-180(appliedphotophysics有限公司)在190-260nm波長范圍內進行紫外掃描分析。圖15顯示了圓二色性色譜分析ifn-lpetggh6、ifn-br、ifn-α、ifn-pmpc的二級結構。通過圓二色性譜分析,均在190-260nm波長范圍內的圓二色譜都呈現出同樣的209/219nm雙峰曲線,且與ifn-α曲線重疊良好,表明在蛋白質上生長原位高分子對蛋白質分子的二級結構沒有明顯影響。用二喹啉甲酸法(bca)測定ifn-pmpc的蛋白濃度,已知濃度的牛血清白蛋白為標準樣,具體步驟按照說明書。上述三制備的ifn-pmpc與上述二制備的ifn-pmpc檢測結果無顯著差異。四、對照方法制備在2.5ml含有40μmifn-α的pbs溶液(溶質為ifn-α,溶劑為ph值為7.4、濃度為10mm的pbs溶液)加入一定量的mpc(mpc與ifn-α的摩爾比分別為1000:1)和11oμl維生素c水溶液(4omg/ml),通氮氣15min后,加入10μl的氯化亞銅和n,n,n`,n'`,n'`一五甲基二乙烯基三胺(pmdta)(濃度分別為100mm和300mm)混合物儲液,密閉條件下反應lh,然后暴露在空氣中終止反應,得到反應混合物。反應混合物經陰離子交換柱(hitrapcaptoqcolumn,gehealthcare)分離純化(純化方法同前)出ifn-pmpc,收集8ml柱體積的為干擾素高分子結合體ifn-pmpc(對照組);將ifn-pmpc(對照組)用sds-page檢測和gpc測定,分子量為59.1kda。計算產率,公式為(得到ifn-pmpc結合體的質量/ifn-lpetggh6的質量)*100%,結果如表3,ifn-pmpc(對照組)的產率為18.9%。表3可以看出,本發明的二的方法與三的對照方法制備的同樣分子量的ifn-pmpc結合體比較,本發明的二的方法制備的ifn-pmpc結合體的產率遠遠高于三的對照方法,說明本發明的方法效果好。實施例2、干擾素高分子結合體ifn-pmpc的功能驗證1、ifn-pmpc的體外生物活性本發明中ifn-pmpc(選擇實施例2的二的方法制備的ifn-pmpc,即分子量為57kda進行表征,下均同)的抗細胞增殖活性采用mtt方法測定。選用了人burkitt’sb淋巴瘤細胞(daudib),因為該細胞對ifn-α2具有較高的敏感。daudib細胞在含有15%fbs、50u/ml盤尼西林和50μg/ml鏈霉素的rmpi-1640中培養一段時間后,在96孔板中接種一定濃度的細胞懸浮液(50μl/孔,7500個細胞),將ifn-lpetggh6、ifn-br、ifn-α、派羅欣和純化后的ifn-pmpc結合體樣品系列稀釋,各50μl加入96孔培養板中,設陰性對照(不含ifn-α2)和空白對照(只含培養液),37℃,5%co2培養72h,加mtt溶解液20μl/孔,3h后用酶標儀測定各孔490nm波長的吸收值,比較不同樣品處理后細胞增殖的程度。圖16顯示了mtt測定ifn-pmpc的體外生物活性,表4顯示了ifn-α、ifn-lpetggh6、ifn-br、派羅欣和ifn-pmpc的體外生物活性,其ic50分別為10.79pg/ml、10.78pg/ml、11.18pg/ml、175.00pg/ml和20.02pg/ml,活性保持率分別為100%、100.09%、96.50%、6.17%和53.90%。綜上可知,ifn-pmpc體外抗腫瘤活性遠高于派羅欣,表明原位atrp沒有嚴重降低ifn的活性,為體內抗腫瘤活性測試提供了依據。表4樣品ifn-αifn-lpetggh6ifn-br派羅欣ifn-pmpcic50(pg/ml)10.7910.7811.18175.0020.02相對活性(%)100100.0996.506.1753.902、ifn-pmpc的藥物代謝動力學測試以下所有動物實驗均在清華大學關于動物實驗的各項規定指導下完成。本發明中利用bal/cnude裸鼠模型經尾靜脈注射ifn-α、派羅欣和ifn-pmpc,測定了血液中干擾素濃度隨時間變化的情況,并利用das軟件進行數據分析。在藥物處理期前,9只雌性裸鼠觀察一段時間后隨機分成3組。以1mg/kg體重劑量尾靜 脈注射ifn-pmpc、派羅欣及未修飾ifn-α對照物,然后在設定的時間點(1,5,15,30min,1,3,6,24,48,72和96h)尾靜脈取血20μl(采血管預先用肝素鈉(江蘇萬邦生化醫藥股份有限公司產品)浸潤并烘干),室溫靜置1h,在4℃、3000×g下離心收集上層血漿,保存于﹣80℃低溫冰箱。用人ifn-α2elisa試劑盒(pblinterferonsource)根據說明書測定血清中的ifn-α2含量。利用das3.0藥物代謝動力學分析軟件計算ifn-pmpc、派羅欣和ifn-α的藥物代謝動力學參數。表5和圖17分別顯示了ifn-pmpc、派羅欣和ifn-α在裸鼠中藥物代謝動力學情況。ifn分布半衰期(t1/2α)消除半衰期(t1/2β)分別為0.42h和1.58h,在給藥5分鐘后,血液中干擾素的濃度即迅速下降到初始劑量的50%以下,24h后殘留濃度不足初始濃度的0.1%。而ifn-pmpc在體內的濃度是逐漸減少的,其初始半衰期和消除半衰期分別為2.04h和51.62h,分別是ifn-α的6.2倍和34.7倍。ifn-pmpc的藥時曲線下面積(auc0-∞)是ifn-α的194.1倍。以上結果表明,與未修飾的ifn-α相比,ifn-pmpc結合體的消除半衰期和體內平均存留時間明顯延長,藥時曲線面積顯著增加,清除率顯著降低,與派羅欣具有相近的體內循環半衰期。表53、ifn-pmpc體內組織分布情況本發明中利用移植了卵巢癌細胞的裸鼠測定了給藥2h,24h和4d后腫瘤中殘留的干擾素的濃度。人卵巢癌細胞(ovcar-3)在含有10%fbs,50u/ml盤尼西林和50μg/ml鏈霉素的rmpi-1640培養基中培養一段時間后,用胰蛋白酶消化剝離,經pbs洗滌,重懸于不含上述添加物的rmpi-1640培養基并與bdmatrigelmatrix等體積混合物中,0.2ml單細胞懸液(5×106個細胞)接種于裸鼠左后肢股骨處背部皮下,培養30天后形成100-200mm3大小的實體瘤腫塊。將裸鼠隨機分成3組,ifn-pmpc、派羅欣、ifn-α以尾靜脈注射打入裸鼠體內,給藥劑量為10μg/20g體重。給藥2h,24h和4d后處死裸鼠,收集心臟、腎臟、肝臟、脾臟、肺臟、胰腺、胃、肌肉、小腸和腫瘤等組織器官。用提取緩沖液(pbs含1mmedta,0.5%tritonx-100,0.5%脫氧膽酸鈉,1mmpmsf,按1:100比例稀釋的蛋白酶抑制劑混合物和磷酸酶抑制劑混合物(sigma-aldrich))破碎后,離心取上清提取液。組織中ifn的濃度用elisa方法定量測定。圖18為ifn-pmpc在各組織中的累積情況,說明ifn-pmpc在心臟、腎臟、肝臟、脾臟、肺臟、胰腺、胃、肌肉、小腸和腫瘤中均能夠有效累積。其中,a圖為ifn-pmpc、派羅欣、ifn-α在腫瘤中累積情況,可以看出,ifn-pmpc組腫瘤中干擾素的濃度和ifn組腫瘤中干擾素的濃度相比,注射2h為12.9倍,注射24h為158.2倍,注射4d后ifn-pmpc仍有20%的殘留。b圖為ifn-pmpc、派羅欣、ifn-α在其他組織中累積的情況。表明,ifn-pmpc能夠有效利用增強通透和滯留效應,使干擾素在腫瘤組織中聚集、在血液循環中半衰期延長,從而提高干擾素在體內的生物利用度和抗腫瘤功效。4、裸鼠模型測試ifn-pmpc體內抗腫瘤活性ifn-pmpc的體內生物活性用動物移植性腫瘤實驗法測定。按上述方法,ovcar-3細胞接種于裸鼠左后肢股骨處背部皮下,培養3周天后形成實體瘤腫塊(~20mm3),從而建立裸鼠腫瘤模型。將26只裸鼠分成4組,ifn-pmpc、派羅欣及對照品(ifn-α為陽性對照,生理鹽水為陰性對照)以尾靜脈注射打入裸鼠體 內,給藥劑量為20μg/20g體重,每周注射一次,直至生理鹽水組、ifn-α組和派羅欣組的裸鼠全部死亡。在本實驗中裸鼠死亡包括自然死亡和安樂處死,安樂處死是指裸鼠腫瘤生長超過500mm3或體重下降超過15%,被注射巴比妥鈉類藥物處死。每周觀察裸鼠生存狀態以及腫瘤生長情況,動態測量裸鼠體重和腫瘤體積隨時間的變化。為檢測ifn-pmpc的毒性,給藥三次后,處死小鼠,并收集腫瘤、心臟、肝臟和腎臟,固定在4%甲醛溶液中,切片后通過標準方法進行he染色,觀察器官的組織學形態。結束治療后,通過眼球取血,測量乳酸脫氫酶、肌酸激酶同工酶、谷丙轉氨酶、谷草轉氨酶、肌酐、尿素氮、紅細胞、白細胞、血小板、血紅蛋白等基本生理指標。實驗結果表明(圖19和圖20),實驗期間,生理鹽水組的裸鼠的腫瘤體積快速增大,注射第42天,裸鼠腫瘤體積均超過500mm3,生存中值僅為35天;實驗期間,ifn-α組的裸鼠的腫瘤體積逐漸增大,注射第49天ifn組的裸鼠腫瘤體積也超過500mm3,生存中值為38天,沒有體現明顯的抗腫瘤活性;實驗期間,派羅欣組的裸鼠的腫瘤體積逐漸增大,注射第63天ifn組的裸鼠腫瘤體積也超過500mm3,生存中值為60天,有一定的抗腫瘤活性;實驗期間,ifn-pmpc組的裸鼠的腫瘤體積基本沒有變化,同時,有75%的小鼠腫瘤消失,1只小鼠腫瘤不再長大,只有1只小鼠腫瘤緩慢增大,直到93天腫瘤體積才超過500mm3。以上數據表明,ifn-pmpc能夠有效地抑制腫瘤的生長,具有非常好的體內抗腫瘤活性,甚至優于目前市售藥品派羅欣。實驗結果還表明,實驗期間,ifn-pmpc組的裸鼠體重略有增加(圖21),表明ifn-pmpc對裸鼠沒有明顯的副作用。圖22為給藥3次后小鼠的腫瘤、心臟、肝臟、腎臟的組織學切片,可以看出,注射ifn-pmpc后,小鼠腫瘤細胞間隙出現空穴,胞漿和細胞核形態不明顯,細胞壞死,大塊細胞膜脫落,與對照組差異明顯。同時,心臟、肝臟和腎臟的細胞形態完整,無明顯細胞壞死,與對照組組織學形態無明顯差異。圖23和圖24分別為結束治療后小鼠的生化和血液指標,與生理鹽水組、ifn-α組和派羅欣組的裸鼠相比,ifn-pmpc組的裸鼠的乳酸脫氫酶、肌酸激酶同工酶、谷丙轉氨酶、谷草轉氨酶、肌酐、尿素氮、紅細胞、白細胞、血小板、血紅蛋白等基本生理指標沒有顯著差別。以上數據表明ifn-pmpc不會對裸鼠體內器官造成明顯毒性。本發明的方法原位atrp合成技術產率高,純化簡單,易于工業化,制備的干擾素-pmpc結合體具有較高的生物活性保存率,顯著延長的半衰期和有效的腫瘤抑制作用,與市售藥物派羅欣體內外活性相比表現出更加優異的功效。atrp合成技術可望取代peg化成為修飾蛋白藥物以提高藥物穩定性、改善藥物代謝動力學和增強治療功效的新方法。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1