一種羧甲基化五味子多糖及其制備方法和應用與流程
本發明涉及一種羧甲基化五味子多糖及其制備方法和應用,屬于多糖改性技術領域。
背景技術:
五味子(Schisandra chinensis(Turcz.)Baill.)屬木蘭科五味子植物的成熟果實,主要的產于我國黑龍江、遼寧、內蒙古等地,是一種常見的滋補性中藥,具有極高的藥用和食用價值,已被列入可用于保健食品的物品名單(陸兔林,吳楊,季德黑.五味子多糖提取分離和藥理作用研究進展.中國中藥雜志,2014,39(4):751-754.)。研究發現五味子多糖作為五味子中主要的活性成分,其含量較高,并具有抗腫瘤、抗氧化、抗衰老、抗疲勞等多種生物活性(徐博,沈楠,趙麗晶.五味子各成分最新研究進展.中國老年保健醫學,2013,11(5):74-74.),且對機體正常細胞和組織的毒性極小,具有較好的開發應用價值。
目前,多糖的結構修飾方法主要有磷酸酯化、羧甲基化、乙酰化等。羧甲基化修飾作為一種主要的化學修飾法,是指向多糖鏈上引入羧甲基,此種修飾方法對多糖的溶解度及電負性均有較重要的影響(江樂明,樊燦梅,聶少平.羧甲基化大粒車前子多糖的制備及其生物活性研究.食品科學,2013,34(22):10-14.)。研究發現,一種堿溶性銀耳多糖,不溶于水,在通過羧甲基化修飾后,其在水中的溶解度提高至36.38mg/mL(吳瓊.羧甲基化堿溶性銀耳多糖的研究.長春大學學報:自然科學版,2009,19(10):66-68.)。2005年,郭祀遠等申請的專利“羧甲基裂褶多糖制備方法及在化妝品和抗腫瘤藥物中應用”(申請號:CN200510101678.X)中,利用氯乙酸、氫氧化鈉等常規試劑對裂褶多糖進行羧甲基修飾,改善了其溶解性能并保持了其良好的流變性能和抗腫瘤活性。2011年,仰榴青等申請的專利“一種羧甲基化黑木耳多糖、粗多糖及其制備方法和應用”(申請號:CN201110219047.3)中,采用異丙醇法對黑木耳多糖進行羧甲基修飾,使其溶解度達到0.6mg/mL,是原糖溶解度的6倍,且其體外抗氧化能力也得到顯著提高。
本發明人在五味子多糖的分離純化、結構表征及抗腫瘤活性的前期研究中發現,五味子多糖的水溶性不理想,制約了其進一步的深入研究及開發應用。因此,本發明人對五味子多糖進行羧甲基化修飾,初步確定其結構特征,提供一種利用響應曲面法優化羧甲基化五味子多糖的工藝,并研究了該羧甲基化五味子多糖對持久性有機污染物PCB126所致的免疫毒性的干預作用。有關五味子多糖羧甲基化衍生物的制備及其工藝優化的研究至今未見國內外文獻報道,也未見相應專利公開。
技術實現要素:
本發明的目的是為了解決五味子多糖的水溶性不理想,制約了其進一步的深入研究及開發應用的缺點,提供了一種羧甲基化五味子多糖及其制備方法和應用。
本發明采用如下技術方案:一種羧甲基化五味子多糖,所述羧甲基化五味子多糖的分子量為1.698×104Da,所述羧甲基化五味子多糖的單糖組成為:甘露糖、葡萄糖和半乳糖,摩爾比為1:44.84:3.71,所述羧甲基化五味子多糖的溶解度為12.24-12.76mg/mL,所述羧甲基化五味子多糖在水溶液中呈聚集狀形成分子鏈,所述分子鏈的厚度為0.1-1nm。
羧甲基化五味子多糖的制備方法,按照下述步驟進行:
(1)五味子粗多糖的制備:將新鮮五味子于55-65℃烘箱內干燥12-24h后進行粉碎,采用石油醚對干燥后的五味子進行回流脫脂2-3次,然后將脫脂后的五味子藥渣以料液比1:10-1:15加入蒸餾水進行水提,提取2-3次得到五味子多糖提取液,五味子多糖提取液經減壓濃縮,采用80%的乙醇醇沉3-4次,,得到的醇沉物經離心后,復溶、揮醇,經截留分子量為3000Da的透析袋透析12-36h,真空干燥2-3h得到五味子粗多糖;
(2)五味子多糖的羧甲基化:將五味子粗多糖溶于異丙醇溶液中,攪拌使其充分溶解,五味子粗多糖和異丙醇料液比為1:30-1:40(g/mL),溶解溫度為20-25℃,攪拌時間為15-20min,攪拌速度為60-240r/min,向溶解后的五味子多糖溶液中加入質量分數為15-20%的NaOH溶液進行堿化,NaOH溶液的加入量為異丙醇體積的20-30%,堿化溫度為20-25℃,堿化時間為1-2h,然后在攪拌狀態下繼續向堿化后的五味子粗多糖反應液中加入氯乙酸進行羧甲基化,氯乙酸加入量為0.005-0.08g/mL,水浴溫度為40-80℃,羧甲基化反應時間為1-8h,待反應液冷卻后調節反應液pH至中性,采用乙醇醇沉,離心后得到醇沉產物,將醇沉產物采用少量蒸餾水復溶后進行透析,透析結束后進行真空干燥得到羧甲基化五味子粗多糖;
(3)純化:將步驟(2)得到的羧甲基化五味子粗多糖溶解于0.05-0.1mol/L的NaCl溶液中,然后緩慢滴加至DEAE-52層析柱中,并采用0.05-0.2mol/L的NaCl溶液進行梯度洗脫,繪制洗脫曲線,根據洗脫曲線分別收取合并洗脫液,得到羧甲基化五味子多糖不同組分,選取0.05mol/L NaCl的洗脫組分合并,經截留分子量為3000Da的透析袋透析、真空干燥后,得到羧甲基化五味子多糖。
進一步的,所述步驟(1)中五味子粗多糖的得率為15.9-19.8%,多糖含量為54.4-65.7%,溶解度為0.08-0.12mg/mL。
進一步的,所述步驟(2)中羧甲基化五味子粗多糖的得率為60.2-86.4%,羧甲基化取代度為0.41-0.71,溶解度為0.46-0.54mg/mL。
進一步的,所述步驟(1)中回流脫脂的溫度為70-80℃,脫脂時間為1-2h,水提溫度為90-100℃,提取時間為4-6h。
進一步的,所述步驟(2)中采用80%乙醇醇沉24-48h,醇沉液以4000-5000rpm轉速離心5-10min。
進一步的,所述步驟(2)中采用經截留分子量為3000Da的透析袋透析首先采用流水透析,最后進行蒸餾水透析,流水透析時間為36-48h,蒸餾水透析時間為24-36h。
進一步的,所述步驟(2)中真空干燥時間為2-3h,溫度為20-25℃,真空度為-0.085--0.1Mpa。
進一步的,所述步驟(3)中羧甲基化五味子粗多糖在NaCl溶液中經震蕩加熱溶解,震蕩水浴的溫度為50-60℃,震蕩頻率為120-160次/分,震蕩溶解的時間10-12h,然后對溶解后的溶液進行離心分離,溶液離心的轉速為:4000-5000r/min,離心時間為8-10min。
進一步的,所述步驟(3)中流水透析時間為36-48h,蒸餾水透析時間為24-36h,真空干燥時間為2-3h,真空干燥溫度為20-25℃,真空干燥真空度為-0.085--0.1MPa。
進一步的,所述羧甲基化五味子多糖能夠作為免疫增強劑用于功能食品和食品添加劑中。
本發明制備方法簡單,步驟易于操作,采用分子修飾的方法制備出羧甲基化五味子粗多糖和羧甲基化五味子多糖;采用本發明中的制備方法得到的羧甲基化五味子多糖的得率和取代度均較高,溶解性好,溶解度分別是原五味子多糖的5倍和125倍,制備得到的羧甲基化五味子多糖能夠有效改善持久性有機污染物PCB126的免疫抑制作用,能夠作為免疫增強劑,應用于功能食品和食品添加劑。
附圖說明
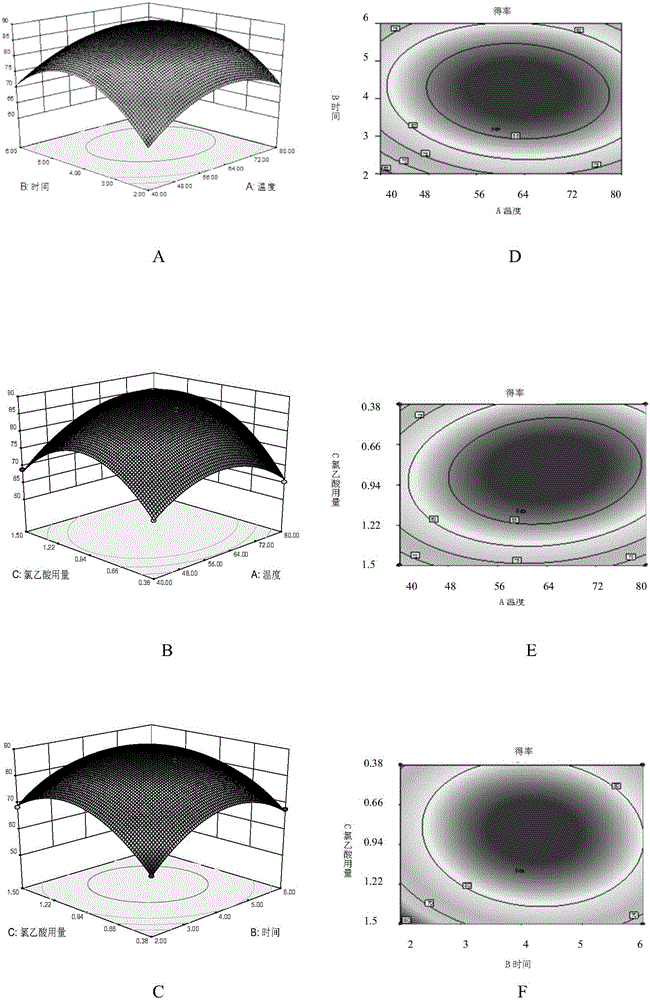
圖1為反應溫度、反應時間、氯乙酸終濃度之間相互作用對羧甲基化五味子多糖得率影響的響應面圖(A、B、C)及等高線圖(D、E、F)。
圖2為反應溫度、反應時間、氯乙酸終濃度之間相互作用對羧甲基化五味子多糖取代度影響的響應面圖(A、B、C)及等高線圖(D、E、F)。
圖3為五味子多糖的紅外光譜圖。
圖4為羧甲基化五味子多糖的紅外光譜圖。
圖5為羧甲基化五味子多糖的原子力二維圖(A)、三維圖(B)和分子鏈高度分布圖(C)。
圖6為羧甲基化五味子多糖對PCB126染毒小鼠脾臟指數的影響圖,ap<0.05,與空白對照組相比;bp<0.05,與模型組相比;cp<0.05,與CSP1高劑量組相比。
圖7為羧甲基化五味子多糖對PCB126染毒小鼠胸腺指數的影響圖,ap<0.05,與空白對照組相比;bp<0.05,與模型組相比;cp<0.05,與CSP1高劑量組相比。
圖8為羧甲基化五味子多糖對PCB126染毒小鼠血清TNF-α水平的影響圖,ap<0.05,與空白對照組相比;bp<0.05,與模型組相比。
圖9為羧甲基化五味子多糖對PCB126染毒小鼠血清INF-γ水平的影響圖,ap<0.05,與空白對照組相比;bp<0.05,與模型組相比。
圖10為羧甲基化五味子多糖對PCB126染毒小鼠血清IL-2水平的影響圖,ap<0.05,與空白對照組相比;bp<0.05,與模型組相比;cp<0.05,與CSP1高劑量組相比。
具體實施方式
五味子粗多糖的制備
實施例1:
將新鮮五味子于55℃烘箱內干燥12h后進行粉碎,采用石油醚對干燥后的五味子于70℃回流脫脂1h,進行2次回流脫脂,然后將脫脂后的五味子藥渣以料液比1:10(g/mL)加入蒸餾水進行水提,于90℃水提4h,提取2次,提取液合并經過減壓濃縮后,80%乙醇反復醇沉3次,離心后,上清液經截留分子量為3000Da的透析袋透析12h、真空干燥2h,制得五味子粗多糖,多糖得率為15.9±0.3%,多糖含量為54.4±2.7%。
實施例2
將新鮮五味子于65℃烘箱內干燥24h后進行粉碎,采用石油醚對干燥后的五味子于80℃回流脫脂2h,進行3次回流脫脂,然后將脫脂后的五味子藥渣以料液比1:15(g/mL)加入蒸餾水進行水提,于100℃水提6h,提取3次,提取液合并經過減壓濃縮后,80%乙醇反復醇沉4次,離心后,上清液經截留分子量為3000Da的透析袋透析36h、真空干燥3h,制得五味子粗多糖,多糖得率為19.8±0.9%,多糖含量為65.7±2.7%,溶解度為0.10±0.02mg/mL。
以實施例2制備的五味子粗多糖為下一步的原料。
與現有文獻中五味子多糖制備工藝相比(趙婷.五味子多糖的分離純化、結構表征及其活性研究[D].江蘇大學,2009.),該粗多糖的制備工藝采用80%乙醇反復醇沉3-4次,顯著提高了五味子粗多糖的多糖含量,多糖含量提高了約21%左右;而采用真空干燥法可有效縮短干燥時間,干燥時間減少了約93%左右,該干燥方法可節約能源,有利于工業生產中的應用。
羧甲基化五味子粗多糖的制備
實施例3:
稱取五味子粗多糖樣品與異丙醇以料液比1:30(g/mL)混合,于20℃下,以轉速為60r/min機械攪拌15min,進行充分溶解;向溶解后的五味子多糖溶液中加入質量分數為15%的NaOH溶液進行堿化,NaOH溶液的加入量為異丙醇體積的20%,溫度20℃下繼續機械攪拌1h;再加入氯乙酸于上述反應液內,使氯乙酸濃度達到0.005g/mL,置于40℃恒溫水浴鍋中,繼續機械攪拌反應1h,冷卻后用鹽酸將反應產物調至中性,經80%乙醇醇沉24h,醇沉液以4000rpm轉速離心5min后,沉淀經復溶,經截留分子量為3000Da的透析袋流水透析36h,蒸餾水透析24h后,于20℃、真空度-0.085MPa下真空干燥2h,即得羧甲基化五味子粗多糖(CSP),多糖得率為75.9±1.1%,羧甲基化取代度為0.41±0.02。
實施例4:
稱取五味子粗多糖樣品與異丙醇以料液比1:35(g/mL)混合,于25℃下,以轉速為240r/min機械攪拌20min,進行充分溶解;向溶解后的五味子多糖溶液中加入質量分數為20%的NaOH溶液進行堿化,NaOH溶液的加入量為異丙醇體積的25%,溫度25℃下繼續機械攪拌2h;再加入氯乙酸于上述反應液內,使氯乙酸濃度達到0.02g/mL,置于60℃恒溫水浴鍋中,繼續機械攪拌反應4h,冷卻后用鹽酸將反應產物調至中性,經80%乙醇醇沉36h,醇沉液以4000rpm轉速離心10min后,沉淀經復溶,經截留分子量為3000Da的透析袋流水透析48h,蒸餾水透析36h后,于25℃、真空度-0.1MPa下真空干燥3h,即得羧甲基化五味子粗多糖(CSP),多糖得率為多糖得率為86.4±1.5%,羧甲基化取代度為0.71±0.03,溶解度為0.50±0.04mg/mL。
實施例5:
稱取五味子粗多糖樣品與異丙醇以料液比1:40(g/mL)混合,于25℃下,以轉速為240r/min機械攪拌20min,進行充分溶解;向溶解后的五味子多糖溶液中加入質量分數為20%的NaOH溶液進行堿化,NaOH溶液的加入量為異丙醇體積的30%,溫度25℃下繼續機械攪拌2h;再加入氯乙酸于上述反應液內,使氯乙酸濃度達到0.08g/mL,置于80℃恒溫水浴鍋中,繼續機械攪拌反應8h,冷卻后用鹽酸將反應產物調至中性,經80%乙醇醇沉48h,醇沉液以5000rpm轉速離心10min后,沉淀經復溶,經截留分子量為3000Da的透析袋流水透析48h,蒸餾水透析36h后,于25℃、真空度-0.1MPa下真空干燥3h,即得羧甲基化五味子粗多糖(CSP),多糖得率為60.2±2.2%,羧甲基化取代度為0.68±0.02。
響應面法優化羧甲基化五味子粗多糖合成工藝
以單因素實驗為基礎,反應溫度選取50℃、60℃和70℃三個水平,反應時間選取2h、4h和6h三個水平,氯乙酸終濃度選取0.01g/mL、0.02g/mL和0.04g/mL三個水平,利用Design Expert 8.0軟件根據Box-Benhnken中心組合設計原理進行實驗設計,以反應溫度(A)、反應時間(B)和氯乙酸終濃度(C)為自變量,以CSP的得率(Y1)和取代度(Y2)為響應值,采用響應曲面法來優化五味子粗多糖羧甲基化工藝條件。響應面實驗因素水平見表1,響應面分析結果見表2、表3及表4。
表1:實驗因素水平表
表2:響應面實驗設計方案及響應值結果
表3得率的方差分析表
注:P<0.0001為極顯著,P<0.05為顯著。
采用Design-Expert 8軟件對實驗數據進行回歸分析,按照各因素對得率試驗結果的影響進行二次方程擬合,擬合得到下式:
Y1=–84.48711+2.53102A+28.50936B+67.94377C-0.036938AB+0.21919AC–1.66175BC–0.020539A2–2.94075B2–36.32919C2
公式中Y1為得率。
各因子對CSP的得率均具有較顯著的影響(P<0.05)。三個因素對于得率的影響順序為:氯乙酸終濃度>反應溫度>反應時間。試驗模型P值<0.0001,證明模型顯著。失擬誤差值不顯著(F值=2.14;P值=0.2374),說明檢驗結果與模型計算結果沒有顯著差異,可用于五味子多糖羧甲基化合成實驗的理論預測。
表4:羧甲基化五味子多糖取代度的方差分析
注:P<0.0001為極顯著,P<0.05為顯著
采用Design-Expert 8軟件對實驗數據進行回歸分析,按照各因素對取代度試驗結果的影響進行二次方程擬合,擬合得到下式:
Y2=-0.68299+0.026417A+0.14229B+0.45287C+4.6875×10-3AB+4.00000×10-3AC–4.84211×10-3BC–2.26187×10-3A2–0.018494B2–0.18924C2
公式中Y2為取代度。
各因子對CSP的取代度具有較顯著的影響(P<0.05)。三個因素對于取代度的影響順序為:氯乙酸終濃度>反應時間>反應溫度。試驗模型的P值<0.0001,證明模型顯著。失擬誤差值不顯著(F值=6.37;P值=0.0528),說明檢驗結果與模型計算結果沒有顯著差異,表明回歸模型與實際情況擬合很好,可用于五味子多糖羧甲基化合成實驗的理論預測。
利用Design Expert 8.0軟件根據多元回歸方程進行繪圖分析,得到回歸方程的響應面圖(見圖1和圖2)三個因素對于CSP的得率和取代度均具有顯著的影響,其中氯乙酸終濃度的影響最顯著。
(5)實驗結果分析與優化:
采用Design-Expert 8軟件對實驗數據進行回歸分析計算,得到合成CSP的最佳工藝條件為:反應溫度為63℃,反應時間為4.5h,氯乙酸終濃度為0.022g/mL。在該條件下,平行實驗5次,得到CSP的得率為88.3±1.65%,取代度為0.72±0.022。實際測定值與響應面模型預測結果較為接近,說明該響應面法優化得到的模型準確可靠,適用于五味子粗多糖羧甲基化的修飾。與2011年,仰榴青等申請的專利“一種羧甲基化黑木耳多糖、粗多糖及其制備方法和應用”(申請號:CN201110219047.3)相比,該工藝條件中氯乙酸用量降低了46%,氯乙酸用量的降低可減少反應過程中多糖降解副反應的發生,使得羧甲基化多糖得率顯著提高,提高了約30%左右;其次,該工藝條件中選用機械攪拌代替磁力攪拌,攪拌強度大,混合效果好,易于調節和操作,更適用于羧甲基化五味子多糖的大量制備;而采用真空干燥法可有效縮短干燥時間,節約能源,有利于工業生產中的應用。
實施例6:
采用響應面得到的最佳工藝參數反應溫度為63℃,反應時間為4.5h,氯乙酸終濃度為0.022g/mL,制備得到羧甲基化五味子粗多糖。
羧甲基化五味子粗多糖的純化及結構特征
稱取上述實施例6中制備羧甲基化五味子粗多糖(CSP)0.5g,進行羧甲基化五味子粗多糖的純化。
實施例7:
溶于0.05mol/LNaCl溶液,于震蕩頻率為160次/分、溫度50℃的震蕩恒溫水浴中充分溶 解10h,溶解液經4000r/min離心10min,除去不溶物后,上清液通過DEAE-52離子交換柱(2.0×50cm)進行分離;以0.05、0.1、0.15和2mol/L的NaCl溶液洗脫,用自動收集器逐管收集洗脫液,將恒流泵的流速控制在1mL/min,每管收集5mL。收集液用硫酸-苯酚法逐管檢測,根據洗脫曲線分別收取合并洗脫液,其中0.05mol/L NaCl的洗脫液經減壓濃縮,截留分子量為3000Da的透析袋流水透析48h、蒸餾水透析24h后,于溫度20℃、真空度為-0.085MPa的真空干燥箱內干燥2h,得到羧甲基化五味子多糖組分(CSP1)。
實施例8:
溶于0.08mol/LNaCl溶液,于震蕩頻率為150次/分、溫度55℃的震蕩恒溫水浴中充分溶解11h,溶解液經4500r/min離心9min,除去不溶物后,上清液通過DEAE-52離子交換柱(2.0×50cm)進行分離;以0.05、0.1、0.15和2mol/L的NaCl溶液洗脫,用自動收集器逐管收集洗脫液,將恒流泵的流速控制在1mL/min,每管收集5mL。收集液用硫酸-苯酚法逐管檢測,根據洗脫曲線分別收取合并洗脫液,其中0.05mol/L NaCl的洗脫液經減壓濃縮,截留分子量為3000Da的透析袋流水透析40h、蒸餾水透析30h后,于溫度21℃、真空度為-0.090MPa的真空干燥箱內干燥2.5h,得到羧甲基化五味子多糖組分(CSP1)。
實施例9:
溶于0.1mol/LNaCl溶液,于震蕩頻率為120次/分、溫度60℃的震蕩恒溫水浴中充分溶解12h,溶解液經5000r/min離心8min,除去不溶物后,上清液通過DEAE-52離子交換柱(2.0×50cm)進行分離;以0.05、0.1、0.15和2mol/L的NaCl溶液洗脫,用自動收集器逐管收集洗脫液,將恒流泵的流速控制在1mL/min,每管收集5mL。收集液用硫酸-苯酚法逐管檢測,根據洗脫曲線分別收取合并洗脫液,其中0.05mol/L NaCl的洗脫液經減壓濃縮,截留分子量為3000Da的透析袋流水透析36h、蒸餾水透析36h后,于溫度25℃、真空度為-0.1MPa的真空干燥箱內干燥3h,得到羧甲基化五味子多糖組分(CSP1)。
與現有文獻(趙婷.五味子多糖的分離純化、結構表征及其活性研究[D].江蘇大學,2009.)和專利(申請號:CN201110219047.3及CN201210132340.0)相比,該純化過程中多糖經恒溫震蕩進行充分溶解,減少了羧甲基化五味子多糖原料的浪費;而柱純化過程采用直徑較大的離子交換柱,使得上樣量增加了一倍,在不改變純化效果的基礎上顯著加快了純化速度;而采用真空干燥法可有效縮短干燥時間,節約能源,有利于工業生產中的應用。
取實施例8制備得到羧甲基化五味子多糖(CSP1),溶解度為12.50mg/mL。
據2005版藥典進行溶解度的測定,五味子粗多糖的溶解度為0.10±0.03mg/mL,羧甲基化五味子粗多糖的溶解度為0.50±0.04mg/mL,羧甲基化五味子多糖(CSP1)的溶解度為12.50 ±0.26mg/mL。
分子量的測定:采用高效凝膠尺寸排阻色譜-多角度激光光散射儀-示差折光檢測儀聯用分析法(HPSEC-MALLS-RI System)對多糖相對分子質量進行測定。色譜條件以TSK-GEL系列G6000PWXL和G4000PWXL色譜柱(300mm,7.8mm,TOSOH,日本)為分析柱,以0.15mol/L NaNO3和0.05mol/L NaH2PO4(pH=7)為流動相,在柱溫為35℃的條件下,以0.5mL/min的流速進行洗脫分析。將CSP1樣品配制成質量濃度為5mg/mL的溶液,經12000r/min的高速離心機離心10min后取上清液過膜后進樣100μL。CSP1分子量為1.698×104Da。與現有文獻(趙婷.五味子多糖的分離純化、結構表征及其活性研究[D].江蘇大學,2009.)和專利(申請號:CN201110219047.3及CN201210132340.0)相比,CSP1分子量較小,而分子量較小的多糖溶解度高且更易于其活性作用的發揮。
單糖組成分析:精密稱取多糖樣品5mg于安瓿瓶中,用2mol/L的硫酸溶液溶解,封口,100℃烘箱中加熱水解8h后,水解液加碳酸鋇中和至中性,以4000r/min離心除去BaSO4沉淀,上清液冷凍干燥。水解產物經乙酰化后用島津GC 2010氣相色譜系統分析。CSP1的單糖組成為:甘露糖、葡萄糖和半乳糖,其摩爾比為1:44.84:3.71。可見CSP1由三種單糖組成,分別為甘露糖、葡萄糖和半乳糖,而現有文獻(趙婷.五味子多糖的分離純化、結構表征及其活性研究[D].江蘇大學,2009.)和專利(CN201210132340.0)中,五味子多糖由六種單糖構成(鼠李糖、阿拉伯糖、木糖、甘露糖、葡萄糖和半乳糖)。
紅外測定:以溴化鉀(KBr)壓片法,取一定量的干燥的多糖樣品,采用傅立葉變換紅外光譜儀(ATAVAR360)掃描4000~400cm-1波長段進行結構表征。五味子多糖經羧甲基化后的多糖于1600cm-1、1420cm-1處均出現強吸收峰,(為-COO-的特征吸收峰,其中1600cm-1左右處為-C-O-的非對稱伸縮振動吸收峰,1420cm-1左右處為-C-O-的對稱伸縮振動吸收峰),表明羧甲基化成功。
原子力電鏡:將多糖樣品配制成1mg/mL的溶液,過膜并收集濾液,取20μL濾液使其均勻附著在干凈的云母片上,室溫下防灰干燥。用掃描探針顯微鏡(MFP-3D)對樣品進行觀測。在水溶液中呈聚集狀,分子鏈的厚度在0.1-1nm。與現有文獻(趙婷.五味子多糖的結構、生物活性及其免疫機制研究[D].江蘇大學,2013.)相比,羧甲基化五味子多糖在水溶液中的聚集減少,且多糖鏈厚度較五味子多糖組分(1-5nm)顯著降低。
羧甲基化五味子多糖對PCB126致機體免疫毒性的干預作用
實驗動物
ICR小鼠(清潔級),雌雄各半,體重為20±2g,購于江蘇大學動物實驗中心,實驗之前檢疫3天,飼養溫度范圍為21±1℃,飼養濕度范圍為60±5%。
材料與試劑
取實施例8純化得到的羧甲基化五味子多糖;
PCB126、生理鹽水、苦味酸;
TNF-α酶聯免疫試劑盒、IL-2酶聯免疫試劑盒、IFN-γ酶聯免疫試劑盒。
實驗分組與給藥
于實驗前適應喂養70只ICR小鼠3天后,隨機分成5組,每組14只,雌雄各半并分開喂養,設置空白對照組、PCB126模型組、羧甲基化五味子多糖組分(CSP1)高、中、低三種濃度劑量組。每天給藥1次,連續14天。而于給藥第7天,PCB126模型組、CSP1高、CSP1中、CSP1低三種濃度劑量組同時灌胃給予50μg/kg PCB126(灌胃量均為每只0.1mL),持續給毒5天。
分組、劑量和給藥途徑如下:
空白對照組:每天灌胃相同劑量的生理鹽水(0.1mL/20g/d),進食和飲水自由;
PCB126模型組:每天灌胃相同劑量的生理鹽水(0.1mL/20g/d),于第7天開始灌胃給予PCB126,給予5天,進食和飲水自由;
CSP1給藥組:分高、中、低三種濃度劑量組,
CSP1高劑量組:每天分別灌胃相同劑量的CSP1(400mg/kg/d),于給藥第7天開始灌胃給予PCB126,給予5天,進食和飲水自由;
CSP1中劑量組:每天分別灌胃相同劑量的CSP1(200mg/kg/d),于給藥第7天開始灌胃給予PCB126,給予5天,進食和飲水自由;
CSP1低劑量組:每天分別灌胃相同劑量的CSP1(100mg/kg/d),于給藥第7天開始灌胃給予PCB126,給予5天,進食和飲水自由。
(1)免疫器官指數
末次給藥后,于次日禁食8h,稱量動物體重并記錄。摘除眼球取血后處死,取出胸腺、脾臟經生理鹽水清洗后用濾紙吸干,稱重并記錄。按下式計算各臟器指數:
(2)血液指標
用ELISA酶聯免疫試劑盒測定小鼠血清中IL-2、INF-γ、TNF-α的含量。
統計學處理
用SPSS16.0統計軟件對數據進行分析。結果表示為組間差異采用單因素方差分析(One-way ANOVA),采用Tukey檢驗法比較各組間差異顯著性,P﹤0.05表示有顯著性差異。實驗結果
CSP1對染毒小鼠的脾臟指數、胸腺指數分別見圖6和圖7。與空白對照組相比,模型組的小鼠脾臟和胸腺指數顯著下降(P<0.05),說明PCB126致小鼠免疫抑制模型建立成功;而CSP1高、中、低劑量均不同程度上恢復了小鼠的脾臟和胸腺指數,其中CSP1高劑量組可同時顯著提高小鼠脾臟指數與胸腺指數。CSP1對染毒小鼠血清中TNF-α、IFN-γ、IL-2水平的影響分別見圖8、圖9和圖10。與空白對照組相比,模型組的小鼠血清中TNF-α、IFN-γ和IL-2水平均顯著下降(P<0.05),而CSP1高、中、低劑量小鼠血清中TNF-α、IFN-γ和IL-2含量均有不同程度的上升,其中CSP1高劑量組小鼠血清中的TNF-α的含量較模型組顯著上升(P<0.05),且CSP1高劑量組和中劑量小鼠血清中IL-2的含量較模型組顯著上升(P<0.05)。目前,現有文獻報道,五味子多糖(趙婷.五味子多糖的結構、生物活性及其免疫機制研究[D].江蘇大學,2013.)與黑木耳多糖(宋廣磊.黑木耳多糖的分離制備及生物活性研究[D].浙江工商大學,2011.)具有免疫調節活性,可顯著改善環磷酰胺所致的免疫抑制作用,但對于持久性有機污染物PCB126所致機體免疫毒性的干預作用未見相關報道。
- 還沒有人留言評論。精彩留言會獲得點贊!