一種銥配合物及其制備方法和應用與流程
本發明涉及發光材料技術領域,特別涉及一種銥配合物及其制備方法和應用。
背景技術:
金屬配合物既具有有機物高熒光量子效率的優點,又具有無機物穩定性好的特點,因此被認為是最有應用前景的一類發光材料。在金屬配合物中,由于重金屬原子(例如鉑,金,銥)的引入,金屬與配體之間產生較強的自旋軌道耦合,使其具有良好的磷光特性。這些豐富的光學特性使重金屬配合物在各種光電器件中得到了廣泛的應用,尤其是銥Ir(III)配合物。由于銥的原子序數較大,可使銥Ir(III)配合物產生很強的自旋軌道耦合,有利于磷光發射,因此具有較高的發光效率,化學和熱穩定性,相對較長的磷光壽命,以及發光顏色可調的特性。
銥配合物根據其結構差異可分為兩類,即具有單一配體的同配體銥配合物和具有兩個相同主配體和一個輔助配體的異配體銥配合物。近年來,隨著對銥配合物的研究日益增多,越來越多的具有聚集誘導磷光(AIPE)特性的銥配合物被報道,這些化合物在溶液狀態不發射但是固體狀態表現出很高的磷光發射,使得它們在有機電致發光領域有著廣闊的前景。
雖然熒光共振能量轉移(FRET)已經被許多小組研究,但是具有FRET特性的銥Ir(III)配合物仍然非常稀少,相比大多數FRET體系,這些配合物往往具有較大的斯托克斯位移。因此,設計并合成具有熒光和磷光雙發射性質的銥Ir(III)配合物成為發光材料領域亟待解決的問題。
技術實現要素:
本發明的目的在于提供一種具有熒光和磷光雙發射性質的銥配合物及其制備方法和應用。
本發明提供了一種銥配合物,具有式Ⅰ通式所示的化學組成:
[(X)2Ir(L2)]Y 式Ⅰ;
其中,X為2-(2,3,4,5-四氟苯基)吡啶或2,4-二氟苯聯吡咯;
L2為3,6-二-叔丁基-9-(4-(4,5-二甲基-2-(吡啶-2-基)-1H-咪唑-1-基)丁基)-9H-咔唑;
Y為一價鹵素陰離子。
優選的,所述一價鹵素陰離子包括PF6-,Cl-,Br-,I-和BF4-中的一種。
本發明提供了一種上述技術方案所述銥配合物的制備方法,包括以下步驟:
(1)將環金屬配體、三氯化銥和混合溶劑混合,加熱反應得到橋氯二聚體,所述環金屬配體為2-(2,3,4,5-四氟苯基)吡啶或2,4-二氟苯聯吡咯;
(2)將所述步驟(1)得到的橋氯二聚體、輔助配體和有機溶劑混合,加熱回流得到銥配合物前驅體,所述輔助配體為3,6-二-叔丁基-9-(4-(4,5-二甲基-2-(吡啶-2-基)-1H-咪唑-1-基)丁基)-9H-咔唑;
(3)將所述步驟(2)中得到的銥配合物前驅體與鹵素鹽混合,取代反應得到銥配合物。
優選的,所述步驟(1)中加熱反應的溫度為140~160℃,加熱反應的時間為20~28h。
優選的,所述步驟(2)中加熱回流的溫度為140~160℃,加熱回流的時間為10~14h。
優選的,所述步驟(3)中取代反應的溫度為15~30℃,取代反應的時間為50~80min。
優選的,所述步驟(3)中的鹵素鹽為包括PF6-,Cl-,Br-,I-或BF4-的鹽。
本發明提供了上述技術方案所述銥配合物或按照上述技術方案所述制備方法制備的銥配合物作為發光材料的應用。
本發明提供了上述技術方案所述銥配合物或按照上述技術方案所述制備方法制備的銥配合物在指紋檢測中的應用。
優選的,所述銥配合物在指紋檢測中的應用包括以下步驟:
將銥配合物溶解,得到混合溶液;
將所述混合溶液涂覆于有指紋的材料表面,靜置后清洗得到待檢測樣品;
將所述待檢測樣品經紫外光照得到指紋圖。
本發明提供了一種銥配合物[(X)2Ir(L2)]Y;其中,X為2-(2,3,4,5-四氟苯基)吡啶或2,4-二氟苯聯吡咯;L2為3,6-二-叔丁基-9-(4-(4,5-二甲基-2-(吡啶-2-基)-1H-咪唑-1-基)丁基)-9H-咔唑;Y為陰離子。本發明提供的銥配合物以2-(2,3,4,5-四氟苯基)吡啶或2,4-二氟苯聯吡咯作為環金屬配體,以3,6-二-叔丁基-9-(4-(4,5-二甲基-2-(吡啶-2-基)-1H-咪唑-1-基)丁基)-9H-咔唑作為輔助配體,由于金屬-配體電荷轉移和配體到配體電荷轉移,使其具有熒光-磷光雙發射,熒光共振能量轉移,聚集誘導熒光發射(AIFE)和聚集誘導磷光發射(AIPE)特性;并且由于輔助配體中的叔丁基以及環金屬配中的氟原子可以與指紋中分泌的化學物質的油脂產生親油脂的吸附作用形成聚集體,使該銥配合物能夠作為染料增強潛在指紋的可視化程度。實驗結果表明,本發明提供的銥配合物可以對不銹鋼尺,玻璃,塑料泡沫和聚丙烯塑料上的指紋進行準確識別,并且能夠對老化程度不同(10天,20天和40天)的指紋進行清楚識別。
附圖說明
圖1為本發明實施例1制備的銥配合物在室溫下在二氯甲烷溶液中的紫外-可見吸收和發射譜圖;
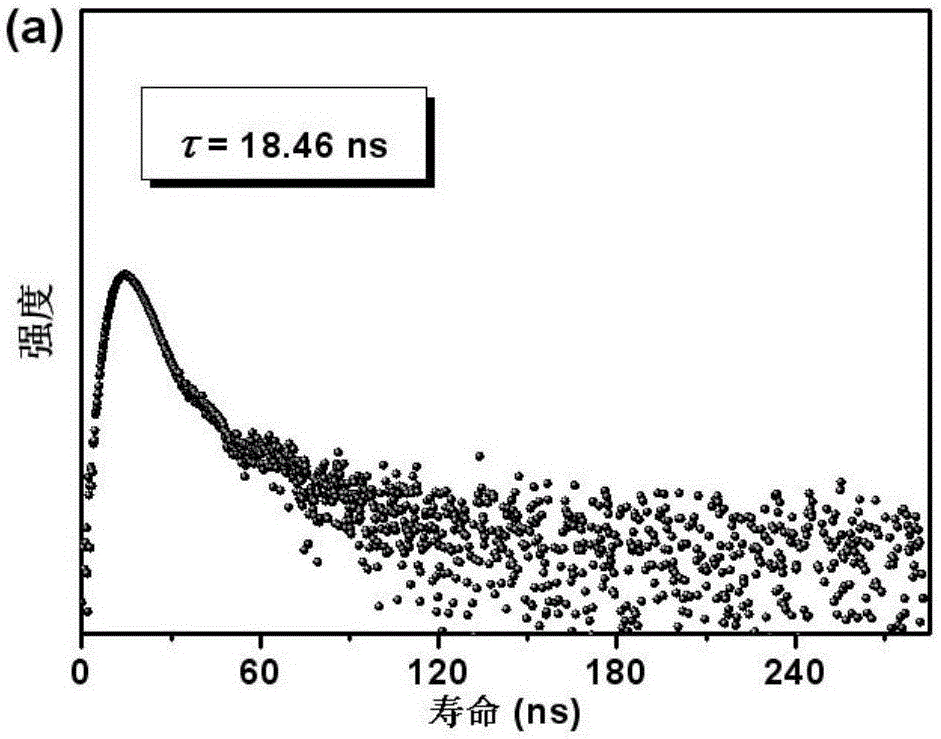
圖2為本發明實施例1制備的銥配合物在熒光區域的衰減曲線;
圖3為本發明實施例1制備的銥配合物在磷光區域的衰減曲線;
圖4為本發明實施例1制備的銥配合物在乙腈-水的混合溶液中伴隨不同水含量(0-90%)的發射譜圖;
圖5為本發明實施例1制備的銥配合物在乙腈/水(3:7)溶液中形貌及粒徑分布圖;
圖6為本發明實施例2的流程圖;
圖7為本發明實施例2中不同基板表面的指紋圖像,其中,(a)為不銹鋼尺,(b)為玻璃,(c)為塑料泡沫,(d)為聚丙烯塑料;
圖8為本發明實施例2中不同志愿者的指紋圖像;
圖9為本發明實施例2中的老化指紋圖,其中,(a)為老化10天,(b)為老化20天,(c)為老化40天;
圖10為本發明對比例1中不同基板表面的指紋圖像,其中,(a)為不銹鋼尺,(b)為玻璃,(c)為塑料泡沫,(d)為聚丙烯塑料。
具體實施方式
本發明提供了一種銥配合物,具有式Ⅰ通式所示的化學組成:
[(X)2Ir(L2)]Y 式Ⅰ;
其中,X為2-(2,3,4,5-四氟苯基)吡啶(tfppy)或2,4-二氟苯聯吡咯(dfppz);
L2為3,6-二-叔丁基-9-(4-(4,5-二甲基-2-(吡啶-2-基)-1H-咪唑-1-基)丁基)-9H-咔唑;
Y為一價鹵素陰離子。
在本發明中,所述所述一價鹵素陰離子包括PF6-,Cl-,Br-,I-和BF4-中的一種。
在本發明中,當所述環金屬配體為2-(2,3,4,5-四氟苯基)吡啶(tfppy)時,所述銥配合物中由于具有更多的氟原子,在指紋識別方面的效果更好,成像更加清晰。
在本發明的實施例中,所述銥配合物可具體為[(tfppy)2Ir(L2)]+PF6-或[(dfppz)2Ir(L2)]+PF6-,其中,L2為3,6-二-叔丁基-9-(4-(4,5-二甲基-2-(吡啶-2-基)-1H-咪唑-1-基)丁基)-9H-咔唑。在本發明中,所述[(tfppy)2Ir(L2)]+PF6-的結構式優選如式Ⅱ所示;所述[(dfppz)2Ir(L2)]+PF6-的結構式優選如式Ⅲ所示。
本發明還提供了一種上述技術方案所述銥配合物的制備方法,包括以下步驟:
(1)將環金屬配體、三氯化銥和混合溶劑混合,加熱反應得到橋氯二聚體,所述環金屬配體為2-(2,3,4,5-四氟苯基)吡啶或2,4-二氟苯聯吡咯;
(2)將所述步驟(1)得到的橋氯二聚體、輔助配體和有機溶劑混合,加熱回流得到銥配合物前驅體,所述輔助配體為3,6-二-叔丁基-9-(4-(4,5-二甲基-2-(吡啶-2-基)-1H-咪唑-1-基)丁基)-9H-咔唑;
(3)將所述步驟(2)中得到的銥配合物前驅體與鹵素鹽混合,取代反應得到銥配合物。
本發明將環金屬配體、三氯化銥和混合溶劑混合,加熱反應得到橋氯二聚體,所述環金屬配體為2-(2,3,4,5-四氟苯基)吡啶或2,4-二氟苯聯吡咯。在本發明中,所述環金屬配體和三氯化銥的摩爾比優選為1.8~2.2:1,更優選為1.9~2:1。
本發明對所述環金屬配體的來源沒有特殊的限定,采用市售產品或本領域技術人員熟知的2-(2,3,4,5-四氟苯基)吡啶或2,4-二氟苯聯吡咯的制備方法制備即可。在本發明中,所述三氯化銥優選為一水合三氯化銥或多水合三氯化銥,更優選為三水合三氯化銥。
在本發明中,所述混合溶劑優選包括醇類和/或醚類溶劑和水。在本發明中,所述醇類和/或醚類溶劑和水的體積比優選為2.5~3.5:1。在本發明中,所述醇類和/或醚類溶劑優選包括2-乙氧基乙醇。
本發明對所述混合溶劑的用量沒有特殊的限定,采用本領域技術人員熟知的反應溶劑的用量即可。在本發明中,所述混合溶劑的體積與所述三氯化銥的物質的量之比優選為15~18L:1mol。
在本發明中,所述加熱反應的溫度優選為140~160℃,更優選為145~155℃;所述加熱反應的時間優選為20~28h,更優選為23~25h。在本發明中,所述加熱反應優選在惰性氣體保護下進行。在本發明中,所述惰性氣體優選為氮氣或氬氣。在本發明中,所述加熱反應過程中,金屬配體與銥鹽反應生成橋氯二聚體。在本發明中,所述橋氯二聚體的通式優選為[Ir(X)2Cl]2,其中X為tfppy或dfppz。
本發明優選在所述加熱反應完成后,將所述加熱反應的產物進行后處理,得到橋氯二聚體。在本發明中,所述后處理優選包括:將所述加熱反應的產物與水混合,得到析出物;將所述析出物過濾后干燥,得到橋氯二聚體。在本發明中,所述水可以降低反應產物在混合溶劑中的溶解度,使橋氯二聚體析出。本發明對所述水的用量沒有特殊的限定,在本發明中,優選加水至析出物的量不再增加為止。
本發明對所述過濾和干燥的操作沒有特殊的限定,采用本領域技術人員熟知的過濾和干燥的技術方案即可。在本發明中,所述過濾優選為抽濾。在本發明中,所述干燥的溫度優選為40~50℃,所述干燥的時間優選為6~10h,更優選為7~8h。
得到橋氯二聚體后,本發明將所述橋氯二聚體與輔助配體和有機溶劑混合,加熱回流得到銥配合物前驅體,所述輔助配體為3,6-二-叔丁基-9-(4-(4,5-二甲基-2-(吡啶-2-基)-1H-咪唑-1-基)丁基)-9H-咔唑。在本發明中,所述氯二聚體和輔助配體的摩爾比優選為2~2.5:1,更優選為2.2~2.4:1。
本發明對所述輔助配體的來源沒有特殊的限定,采用市售產品或本領域技術人員熟知的3,6-二-叔丁基-9-(4-(4,5-二甲基-2-(吡啶-2-基)-1H-咪唑-1-基)丁基)-9H-咔唑的制備方法制備即可。在本發明中,所述輔助配體優選按照G.G.Shan,H.B.Li,H.Z.Sun,D.X.Zhu,H.T.Cao andZ.M.Su,J.Mater.Chem.C,2013,1,1440.中記載的技術方案制備得到。
本發明對所述有機溶劑的種類沒有特殊的限定,采用本領域技術人員熟知的有機溶劑即可。在本發明中,所述有機溶劑優選包括醇類和/或醚類溶劑,更優選包括乙二醇乙醚、無水乙醇和乙二醇甲醚中的一種或多種。本發明對所述有機溶劑的用量沒有特殊的限定,采用本領域技術人員熟知的反應溶劑的用量即可。在本發明中,所述有機溶劑的體積與所述輔助配體的物質的量之比優選為40~60L:1mol,更優選為45~55L:1mol。
在本發明中,所述加熱回流的溫度優選為140~160℃,更優選為145~155℃;加熱回流的時間優選為10~14h,更優選為11~13h。在本發明中,所述加熱回流優選在惰性氣體保護下進行。在本發明中,所述惰性氣體優選為氮氣或氬氣。
得到銥配合物前驅體后,本發明將所述銥配合物前驅體與鹵素鹽混合,反應得到銥配合物。在本發明中,所述銥配合物前驅體和鹵素鹽的摩爾比優選為1:5~10,更優選為1:6~8。在本發明中,所述鹵素鹽優選為包括PF6-,Cl-,Br-,I-或BF4-的鹽,更優選為KPF6,KCl,KBr,KI和KBF4中的一種。
本發明優選將所述加熱回流的產物與鹵素鹽混合,取代反應得到銥配合物。在本發明中,所述混合優選在攪拌條件下進行;所述攪拌的速率優選為100~200rpm,更優選為140~160rpm,所述攪拌的時間優選為50~70min。在本發明中,所述取代反應的溫度優選為15~30℃,更優選為20~25℃;所述取代反應的時間優選為50~80min,更優選為60~70min。
取代反應完成后,本發明優選將所述取代反應的產物進行提純,得到銥配合物。本發明對所述提純的操作沒有特殊的限定,采用本領域技術人員熟知的提純的技術方案即可。在本發明中,所述提純優選包括萃取、洗滌和柱層析色譜分離。
在本發明中,所述萃取的萃取劑優選包括二氯甲烷、氯甲烷、乙酸乙酯和乙醚中的一種;所述萃取劑的用量優選為18~22mL/次;所述萃取的目標物質在有機相。在本發明中,所述洗滌的洗滌劑優選包括二氯甲烷和/或水;所述洗滌的次數優選為3~5次。在本發明中,所述柱層析色譜分離優選為硅膠柱層析;所述硅膠的粒徑優選為200~300目;所述柱層析色譜分離的洗脫劑優選包括二氯甲烷和正己烷;所述二氯甲烷和正己烷的體積比優選為1~3:1;所述洗脫的速率優選為0.1~0.3mL/s。
本發明還提供了一種上述技術方案所述銥配合物或按照上述技術方案所述制備方法制備的銥配合物作為發光材料的應用。在本發明中,所述發光材料優選包括熒光材料和磷光材料。
本發明還提供了一種上述技術方案所述銥配合物或按照上述技術方案所述制備方法制備的銥配合物在指紋檢測中的應用。在本發明中,所述應用優選包括以下步驟:
將銥配合物溶解,得到混合溶液;
將所述混合溶液涂覆于有指紋的材料表面,靜置后清洗得到待檢測樣品;
將所述待檢測樣品經紫外光照得到指紋圖。
在本發明中,用于溶解所述銥配合物的溶劑優選為包括乙腈和水的混合溶劑;所述混合溶劑中乙腈和水的體積比優選為0.1~9:1,更優選為0.2~5:1,最優選為0.8~2:1。在本發明中,所述混合溶液中銥配合物的濃度優選為1.0×10-4~1.0×10-5mol/L。
得到混合溶液后,本發明優選將所述混合溶液涂覆于有指紋的材料表面,靜置后清洗得到待檢測樣品。本發明對所述混合溶液的涂覆量沒有特殊的限定,采用本領域技術人員熟知的涂覆量即可。在本發明中所述混合溶液的涂覆量優選為能夠覆蓋整個指紋。在本發明中,所述靜置的時間優選為8~12min。在本發明中,所述清洗的洗滌劑優選為乙腈、四氫呋喃、乙醇或甲醇;所述清洗的次數優選為5~6次。本發明優選在所述清洗完成后進行自然晾干得到待檢測樣品。
本發明優選將所述待檢測樣品經紫外光照得到指紋圖。在本發明中,所述紫外光的波長優選為360~370nm。
為了進一步說明本發明,下面結合實施例對本發明提供的銥配合物及其制備方法和在指紋檢測中的應用進行詳細地描述,但不能將它們理解為對本發明保護范圍的限定。
實施例1:
(1)2-(4,5-二甲基-1H-咪唑-2-基)吡啶(pydmi)的合成
將1.02g(9.5mmol)的吡啶甲醛,0.90g(10.2mmol)的丁二酮,5.50mL乙醇依次加入50mL二口圓底燒瓶,氮氣保護,將1.48g(19.22mmol)的醋酸銨溶于7.04mL乙醇中,然后緩慢滴加到體系中,使其在室溫下攪拌約30min,放于75℃油浴中攪拌回流約24h。
待反應完全,將得到的混合物先以水洗,再用二氯甲烷萃取水層三次,調節水層pH=8,合并有機層,用無水硫酸鈉干燥后旋蒸,得到黑色粘稠液體。將得到的黑色粘稠液體制沙,柱層析分離提純,先用石油醚加壓沖掉紅色物質,再用石油醚:二氯甲烷為10:1洗脫,最后用二氯甲烷沖下來,旋蒸得到暗紅色液體。制沙后進行第二次柱色譜分離,用石油醚:二氯甲烷為10:1洗掉前段雜質,再用二氯甲烷洗脫,除去后段雜質,旋干,得到淡黃色固體,用正己烷:二氯甲烷為4:1重結晶三次,得到0.74g白色固體,產率為45%。
以氘代氯仿(CDCl3)或氘代二甲基亞砜(DMSO-d6)為溶劑使用Bruker Avance 400MHz測得核磁共振氫譜(1HNMR):1HNMR(400MHz,CDCl3,ppm)δ10.40(s,1H),8.45(d,J=5.0Hz),8.06(dt,J1=8.1Hz,J2=1.8Hz,2H),7.70(dt,J1=8.1Hz,J2=1.8Hz,2H),7.15,(ddd,J1=1.2Hz,J2=5.0Hz,J3=7.5Hz,2H),2.19(s,6H).
(2)2-(1-(4-溴丁基)-4,5-二甲基-2-咪唑基)吡啶(Pydmi-C4-Br)的合成
將4.382g(182.6mmol)的氫化鈉加入到100mL三口燒瓶中,氮氣保護,取40mL的四氫呋喃緩慢注入體系。再量取20mL的四氫呋喃將稱取的3.16g(18.26mmol)Pydmi完全溶解,然后用進樣器緩慢加入體系當中(該反應產生大量氫氣且劇烈放熱,整個加入過程在冰水浴中進行),觀察到反應液變為粉紅色,產生大量氣泡,滴加完畢后,使反應在室溫下持續攪拌1h,再將15.6g(73.02mmol)的1,4-二溴丁烷也注入體系,緩慢升溫至70℃,觀察到體系的顏色加深,繼續反應約20h。反應結束后冷卻至室溫,用80mL二氯甲烷萃取,有機相旋干制沙,再進行柱層析分離(石油醚:二氯甲烷=20:1,15:1,10:1,8:1,6:1,3:1),旋蒸后得到2.7g白色固體,產率為48%。
以氘代氯仿(CDCl3)或氘代二甲基亞砜(DMSO-d6)為溶劑使用Bruker Avance 400MHz測得核磁共振氫譜(1HNMR):1HNMR(400MHz,CDCl3,ppm)δ8.54-8.52(m,1H),8.14-8.11(m,1H),7.72-7.68(m,1H),7.17-7.14(m,1H),4.53(t,J=14.0Hz,2H),4.41(t,J=12.4Hz,2H),2.23(s,3H),2.21(s,3H),1.91-1.88(m,4H).
(3)輔助配體(Py-C4-Cz)的合成
將400mg(1.30mmol)的pydmi-C4-Br,365mg(6.50mmol)的氫氧化鉀,41.9mg(0.13mmol)的四丁基溴化銨依次加入到50mL二口圓底燒瓶中,氮氣保護,然后量取15mL的甲苯作為溶劑,將稱取的436mg(1.56mmol)的n-But-Cz完全溶解于其中,再將它和5mL的水依次緩慢注入到體系中。滴加完畢后緩慢升溫至105℃,可以觀察到體系的顏色逐漸變為黑色,繼續反應約20h。待反應完畢后冷卻至室溫,先用30mL的水洗,再用二氯甲烷萃取三次,合并有機相旋干,制沙,進行柱層析分離(石油醚:二氯甲烷=30:1,25:1,20:1,15:1,10:1,8:1,6:1,4:1),最終得到316mg白色固體,產率為48%。
以氘代氯仿(CDCl3)或氘代二甲基亞砜(DMSO-d6)為溶劑使用Bruker Avance 400MHz測得核磁共振氫譜(1HNMR):1HNMR(400MHz,CDCl3,ppm)δ8.39-8.38(m,1H),8.11(d,J=8.0Hz,1H),8.09(d,J=1.6Hz,1H),7.70-7.66(m,1H),7.50-7.47(m,2H),7.24(d,J=3.2Hz,1H),7.13-7.10(m,1H),4.46(t,J=7.6Hz,2H),4.25(t,J=6.8Hz,2H),2.21(s,3H),2.08(s,3H),1.94-1.77(m,4H),1.46(s,18H).
(4)四氟苯基吡啶二氯橋配合物[Ir(tfppy)2CI]2的合成
將812mg(4.25mmol)的2-(3,5-四氟苯基)吡啶,712mg(2.02mmol)的水合三氯化銥依次加入到50mL的二口圓底燒瓶中,氮氣保護,再量取25mL的2-乙氧基乙醇和8mL的水也一并加入體系,緩慢升溫至150℃,反應約24h。待反應完畢先冷卻至室溫,再將反應后的混合液從二口燒瓶轉移至燒杯,在加入適量水后繼續攪拌,可觀察到燒杯中析出大量的黃色固體,攪拌一段時間之后進行抽濾,直至完全干燥,最終得到590mg黃色固體,產率為49%。
(5)[(tfppy)2Ir(L2)]+(PF6-)的合成
配體L2(0.10g,0.20mmol)與橋氯二聚體[Ir(dfppy)2Cl]2(0.11g,0.09mmol)溶于乙10mL乙二醇乙醚溶劑中氮氣保護下在150℃回流12小時反應液冷卻至室溫后,加入KPF6后繼續攪拌約1小時。萃取并洗掉鹽。柱層析色譜分離,二氯甲烷/正己烷作為洗脫劑,用硅膠柱層析純化后得到(1:1,v/v)。獲得綠色固體,產量80%。
以氘代氯仿(CDCl3)或氘代二甲基亞砜(DMSO-d6)為溶劑使用BrukerAvance 400MHz測得核磁共振氫譜(1HNMR):1HNMR(400MHz,DMSO-d6,ppm)δ8.27-8.22(m,3H),8.18(d,J=1.4Hz,2H),8.09-8.01(m,2H),7.95(t,J=2.0Hz,2H),7.77(d,J=5.6Hz,1H),7.53(d,J=5.2Hz,1H),7.48-7.37(m,5H),7.21-7.14(m,2H),4.61-4.44(m,2H),4.36-4.23(m,2H),2.13(s,3H),1.84-1.76(m,4H),1.46(s,3H),1.40(s,18H).
在Vario El III型元素分析儀上進行元素分析(C,H和N):Anal.calcd for IrC56H50N6F14P:C 51.70,H 3.85,N 6.46;found:C 51.73,H 3.87,N 6.41%.
采用voyager matrix時間飛行質譜儀進行高分辨質譜(HRMS)測定:HRMS(ESI-TOF)(M-PF6-):m/z calcd 1151.3598;found:1151.3637.
上述檢測結果表明,本實施例中銥配合物結構如式Ⅱ所示。
用ShimadzuMultiSpec-1501型島津分光光度計和日立F-4600型分子熒光儀測定本實施例中銥配合物在二氯甲烷溶液中的紫外-可見吸收光譜和光致發光光譜,如圖1所示,詳細光物理數據如表1所示。從圖1中可以得出,配合物的主要吸收帶在300nm之前主要歸屬于自旋允許的配體的1π-π*躍遷;而300nm至400nm之間能量較低,能觀測到弱的吸收帶,這主要歸屬于自旋允許以及自旋禁阻的金屬-配體電荷轉移(1MLCT)和配體到配體電荷轉移(1LLCT)躍遷;在450nm之后長長的尾部吸收可以分配到自旋禁阻的3MLCT/3LLCT或3π-π*的躍遷。
表1實施例1中銥配合物在溶液中和在固態的光物理數據
a 298K時在二氯甲烷溶液(1.0×10-5M)中測定。b通過以色氨酸(ФPL=0.14的水,pH=7.2,25℃)和硫酸奎寧(ФPL=0.54在0.1M的H2SO4溶液中)作為參比物測定。c絕對磷光量子產率用校準積分球系統測定。
利用355納秒激光作為激發光源,利用愛丁堡LP980激光閃光光解光譜儀收集本實施例制備的銥配合物發射壽命,得到在熒光和磷光區域的衰減曲線分別如圖2和圖3所示。從圖2可以看出,18.46ns的熒光壽命的迅速衰減,表現出一個典型的正常熒光。相比之下,如圖3所示,發射帶呈現雙指數衰減,也就是說,兩種慢的組分發光壽命為1.61μs和9.40μs,這是由于三線態伴隨著ILCT和MLCT。
上述檢測結果表明,銥Ir(III)配合物的發射特性為熒光-磷光雙發射。熒光是由以配體中心(LC)的π-π*發射態引起的,而磷光光譜所具有的廣泛和普通的特征結構歸因于MLCT/LLCT的特性。另一方面,由于吸收光譜和熒光光譜強烈的重疊,在光物理過程中存在著熒光共振能量轉移。重疊表現在配體(tfppy)或雙環二氯橋聯銥配合物的吸收光譜和熒光光譜之間,從而表明[(tfppy)2Ir(L2)]+PF6-熒光共振能量轉移過程的可能性。熒光共振能量轉移過程的可能性是在同一個配合物中經過放射能量從一個tfppy轉移到另一個tfppy。
利用EdinburghLFS-920型熒光光譜儀測試本實施例中銥配合物的熒光發射光譜,得到本實施例中銥配合物在不同比例的乙腈-水混合溶液中的發射圖譜如圖4所示。從圖4可以看出,因為伴隨著水的不斷加入磷光發光強度持續增加了16倍(從88到1388),說明銥配合物表現出明顯的AIPE特性。隨著水的加入,由于分子間的相互作用和非晶態的形成,能觀察到配合物的發射光譜紅移3至4nm。類似的性質在熒光的短波范圍內表現出明顯的AIFE特性。此外,[(tfppy)2Ir(L2)]+PF6-的AIE特性產生于3ILCT的性質和分子內旋轉受限(RIR)的機理。因此,基于分子間相互作用(C–H…π,C–F…π,C–H…F,C–H…N,等)在聚集狀態,這種配合物由于更多的-F被修飾到配體上,所以比其它具有AIE特性的銥配合物表現出更強烈的發光。
利用FEI Tecnal G2S-Twin測得本發明實施例中銥配合物在混合溶液(70%的水和30%的乙腈含配合物,c=1×10-5M)中的透射電子顯微鏡(TEM)圖以及通過ZetasizerNano ZS90測得動態光散射(DLS)大小如圖5所示。可以看出,銥配合物聚集狀態形成納米顆粒結構,隨著水的加入,在乙腈-水的混合溶液中能觀察到球形聚集體,其平均粒徑約為50nm。
實施例2:
將實施例1制備的銥配合物用于指紋檢測,檢測流程圖如圖6所示。在材料(不銹鋼尺,玻璃,塑料泡沫和聚丙烯塑料)表面印入油脂性指紋,用滴管逐滴將含有銥配合物的乙腈/水的混合溶液(1:9-9:1,v/v,c=1.0×10-4-1.0×10-5M)滴在指紋上面,直至覆蓋整個指紋。約10分鐘后,指紋被仔細地清洗5到6次,然后在空氣中自然干燥。最后,在365nm紫外光照下觀察得到指紋圖如圖7所示。
不銹鋼尺,玻璃,塑料泡沫和聚丙烯塑料表面指紋圖分別如圖7(a)~(d)所示,通過覆蓋樣品溶液約10分鐘得到四個清晰的指紋圖像。第一級(整體凸起的脊狀紋路)以及第二級(中斷、分叉、交叉的脊狀紋路)的細節可以看的很清楚,甚至一些三級信息沿脊狀紋路上的汗毛孔也可以觀察到,這些結果可以對指紋準確識別。
為了研究本方法的可重復性,按照上述方法對幾個志愿者指紋進行采集,結果如圖8所示。可以看出圖像仍然是可高度重復的。
按照上述方法分別對老化10天,20天和40天的指紋進行檢測,結果如圖9所示。結果表明,老化程度不同的指紋依然可以清楚識別。
上述結果表明,本發明提供的銥配合物對指紋的顯像能力強,可以作為指紋識別方面良好又實用的染料。
對比例1:
在材料(不銹鋼尺,玻璃,塑料泡沫和聚丙烯塑料)表面印入油脂性指紋,在可見光下觀察指紋如圖10所示。圖10(a)~(d)分別顯示了不同材料表面的指紋在可見光照射下的圖像,可以看出,圖像的細節是不清楚,難以通過肉眼區分。
實施例3:
(1)2,4-二氟苯聯吡咯(dfppz)的合成
將5.71g(34.80mmol)的1,3,3-四甲氧基丙烷溶于50mL乙醇溶液,攪拌下注入約3mL鹽酸溶液。向上述溶液緩慢滴加5.0g(31.63mmol)2,4-二氟苯肼鹽酸鹽的乙醇溶液100mL。滴加完畢,加熱回流約24小時。
反應結束后,將得到的混合物先以水洗,再用二氯甲烷萃取水層三次,合并有機層,用無水硫酸鈉干燥后旋蒸,得到黑色粘稠液體。將得到的黑色粘稠液體制沙,柱層析分離提純,用石油醚:二氯甲烷為10:1洗脫,旋干,得到無色油狀液體,產率為75%。
以氘代氯仿(CDCl3)或氘代二甲基亞砜(DMSO-d6)為溶劑使用BrukerAvance 400MHz測得核磁共振氫譜(1HNMR):1HNMR(400MHz,CDCl3,ppm)δ7.92(t,J=2.8Hz,1H),7.88-7.82(m,1H),7.73(s,1H),7.02-6.95(m,2H),6.47(t,J=2.1Hz,1H).
(2)二氟苯聯吡咯二氯橋配合物[Ir(dfppz)2CI]2的合成
將812mg(4.25mmol)的2-(3,5-四氟苯基)吡啶,712mg(2.02mmol)的水合三氯化銥依次加入到50mL的二口圓底燒瓶中,氮氣保護,再量取25mL的2-乙氧基乙醇和8mL的水也一并加入體系,緩慢升溫至150℃,反應約24h。待反應完畢先冷卻至室溫,再將反應后的混合液從二口燒瓶轉移至燒杯,在加入適量水后繼續攪拌,可觀察到燒杯中析出大量的黃色固體,攪拌一段時間之后進行抽濾,直至完全干燥,最終得到500mg黃色固體,產率為42%。
(3)[(dfppz)2Ir(L2)]+(PF6-)的合成
將按照實施例1所述方法制備的配體L2(0.10g,0.20mmol)與橋氯二聚體[Ir(dfppz)2Cl]2(0.11g,0.09mmol)溶于乙10mL乙二醇乙醚溶劑中氮氣保護下在150℃回流12小時反應液冷卻至室溫后,加入KPF6后繼續攪拌約1小時。
萃取并洗掉鹽。柱層析色譜分離,二氯甲烷/正己烷作為洗脫劑,用硅膠柱層析純化后得到(3:1,v/v)。獲得綠色固體,產量65%。
從以上對比例和實施例可以看出,本發明提供的銥配合物具有熒光-磷光雙發射,熒光共振能量轉移,聚集誘導熒光發射(AIFE)和聚集誘導磷光發射(AIPE)特性,能夠作為染料增強潛在指紋的可視化程度,對不同材料上的指紋進行準確識別,并且能夠對老化程度不同(10天,20天和40天)的指紋進行清楚識別。
以上所述僅是本發明的優選實施方式,并非對本發明作任何形式上的限制。應當指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!