GC?MS/MS測定食用香精中3?乙酰基?2,5?二甲基噻吩的方法與流程
本發明屬于食品添加劑檢測技術領域,具體涉及gc-ms/ms測定食用香精中3-乙酰基-2,5-二甲基噻吩的方法。
背景技術:
合成香料3-乙酰基-2,5-二甲基噻吩具有燒烤、堅果的氣味,于2002年通過世界衛生組織和聯合國糧農組織下設的食品添加劑聯合專家委員會的安全評估,開始作為食品用香料使用。目前,我國允許其作為食品用合成香料使用,收錄于國標gb2760-2014中。2013年6月4日,歐洲食品安全管理局公布了對該物質重新評估的意見,認為其在體內和體外試驗均表現出致突變性,作為食品用香料使用存在安全問題。2013年6月14日,歐盟委員會通過法規(regulation(ec)no545/2013),禁止該物質作為食品用香料使用,將其從食品用香料法規附錄ⅰ食品用香料清單中刪除。中國香精香料化妝品工業協會針對這一情況迅速做出反應,于2013年8月14日發布自律文件(香化協字[2013]62號),要求香料生產企業不得生產用作食品用香料的3-乙酰基-2,5-二甲基噻吩,食品用香精生產企業調整配方,不得使用該物質。
目前對3-乙酰基-2,5-二甲基噻吩的檢測在國標中尚屬空白,國內針對此物質的定量檢測研究開展的也不多,僅有一篇專利報道以氣相色譜-質譜聯用儀測定,公開號為:cn1046557775a,其公開了選擇性離子監測(sim)模式檢測,內標法進行定量。該法存在的主要缺陷是:由于食用香精成分復雜,通常含結構相近化合物,各組分在檢測中并不一定能完全分離,容易相互干擾,采用單極質譜檢測,準確度和靈敏度都有待考證。
技術實現要素:
針對現有技術存在的問題,本發明的目的在于設計提供一種gc-ms/ms測定食用香精中3-乙酰基-2,5-二甲基噻吩的方法。本發明與單級質譜sim模式相比,本發明中采用多級質譜mrm模式能更準確的定性,同時,因為從干擾嚴重的一級碎片中抽取了母離子,進一步碎裂得到子離子來定量,靈敏度也高于單級質譜,更適合于微量、痕量物質的檢測。
所述的gc-ms/ms測定食用香精中3-乙酰基-2,5-二甲基噻吩的方法,其特征在于包括以下步驟:
1)將食用香精香料樣品用提取溶劑進行超聲提取,冷卻后用提取溶劑定容,混勻后靜置,取上部溶液過微孔濾膜,所述的提取溶劑為正己烷或正庚烷;
2)取步驟1)得到的濾液,采用gc-ms/ms進行測定,外標法定量。
所述的gc-ms/ms測定食用香精中3-乙酰基-2,5-二甲基噻吩的方法,其特征在于所述的步驟1)中超聲提取時間至少為15min。
所述的gc-ms/ms測定食用香精中3-乙酰基-2,5-二甲基噻吩的方法,其特征在于所述的步驟1)中微孔濾膜為有機濾膜,孔徑為0.22μm或0.45μm。
所述的gc-ms/ms測定食用香精中3-乙酰基-2,5-二甲基噻吩的方法,其特征在于所述的步驟2)中gc-ms/ms的色譜條件為:
色譜柱:hp-innowax或相當者;
進樣口溫度:240℃;
程序升溫條件:50℃保持1min,20℃/min升至200℃,保持3min,再25℃/min升至240℃,保持7min;
分流比為10:1;
柱流速為1.0ml/min;
進樣量為1.0μl;
gc-ms/ms質譜條件為:
離子源:電子轟擊源;
電子電離能量:70ev;
離子源溫度:230℃;
四級桿溫度:150℃;
碰撞氣體(n2)流速:1.5ml/min;
淬滅氣體(he)流速:2.25ml/min;
溶劑延遲:8min;
掃描方式:mrm;
母離子、子離子及碰撞能量如表1:
表1母離子、子離子及碰撞能量表(*為定量離子)
所述的gc-ms/ms測定食用香精中3-乙酰基-2,5-二甲基噻吩的方法,其特征在于所述的步驟2)中外標法定量為用提取溶劑溶解3-乙酰基-2,5-二甲基噻吩,并用提取溶劑稀釋至所需濃度,上機測定,以峰面積或峰高繪制標曲。
所述的gc-ms/ms測定食用香精中3-乙酰基-2,5-二甲基噻吩的方法,其特征在于所述的色譜柱規格為30m×0.25mm×0.25μm或相當者。
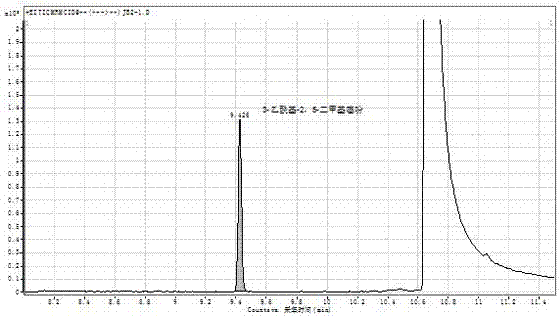
本發明首次采用gc-ms/ms檢測食用香精中3-乙酰基-2,5-二甲基噻吩;采用mrm模式能有效消除基質效應,提高靈敏度和準確度;外標法線性良好,定量準確,檢出限為0.1mg/kg,如圖3所示。本方法前處理簡單,結果可靠,靈敏度高,對監測食用香精中3-乙酰基-2,5-二甲基噻吩的使用情況,保護消費者健康具有一定的現實意義。
附圖說明
圖1是標準品典型圖譜;
圖2是加標回收試驗樣品中3-乙酰基-2,5-二甲基噻吩的典型圖譜;
圖3是檢出限(加標量0.1mg/kg)圖譜。
具體實施方式
下面結合附圖對本發明的技術方案做進一步的說明,但并不局限于此,凡是對本發明的技術方案進行修改或等同替換,而不脫離本發明技術方案的精神和范圍,均應涵蓋在本發明的保護范圍中。
實施例:本發明實施例中的樣品為粉末狀肉味香精
一、標準儲備液的制備
精確稱取3-乙酰基-2,5-二甲基噻吩標準品,用正己烷或正庚烷溶解并定容,配制成濃度為1mg/ml的標準儲備液。
二、線性標準溶液的制備
取1mg/ml的標準儲備液100.0μl,用正己烷或正庚烷稀釋并定容至10ml,得10μg/ml的標準使用液。
取10μg/ml的標準使用液10.0μl、20.0μl、40.0μl、80.0μl、100.0μl,150.0μl、200.0μl、0.25ml,用正己烷分別稀釋并定容至10ml,得濃度為10ng/ml、20ng/ml、40ng/ml、80ng/ml、100ng/ml、150ng/ml、200ng/ml、250ng/ml的線性標準溶液,供gc-ms/ms測定。
三、氣相色譜/質譜條件
色譜條件:采用agilenthp-innowax色譜柱進行分離;進樣口溫度為240℃;程序升溫條件為50℃保持1min,20℃/min升至200℃,保持3min,再25℃/min升至240℃,保持7min;分流比為10:1;柱流速為1.0ml/min;進樣量為1.0μl。
質譜條件:離子源為電子轟擊源;電子電離能量為70ev;離子源溫度為230℃;四級桿溫度為150℃;碰撞氣體(n2)流速為1.5ml/min;淬滅氣體(he)流速為2.25ml/min;溶劑延遲時間為8min;掃描方式為mrm;母離子、子離子及碰撞能量如下表所示。
四、樣品測定
準確稱取樣品0.1g于10ml容量瓶中,加入約7ml正己烷,超聲提取15min,靜置冷卻后用正己烷定容,混勻后靜置數分鐘,上部提取液過0.22μm有機相濾膜,取續濾液待上機測定。同時做試劑空白。
經測試發現,試劑空白無干擾,樣品未檢出。
五、加標回收試驗樣品測定
準確稱取樣品0.1g于10ml容量瓶中,按表2中所列的加標量進行加標,然后同樣品處理,續濾液待上機測定。考察回收率。各加標水平平行測定3份。
表2三水加標試驗加標量及平均回收率表
從表2可知,樣品的加標回收率在94.9~100.3之間,說明本方法回收率較好。
圖1是標準品典型圖譜;圖2是加標回收試驗樣品中3-乙酰基-2,5-二甲基噻吩的典型圖譜。從圖中可以得到,目標物峰型對稱,響應良好。
六、方法精密度試驗
上述加標(中水平)樣品平行制備6份,測定結果見表3。
表3方法精密度試驗結果表
從表3可知,本方法重復性好。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種具有上轉換發光可調的9-乙酰基蒽共晶材料及其制備方法
- N-苯磺酰基-3-乙酰基吲哚酰腙類化合物、制備方法及應用
- N-苯磺酰基-3-(2-基-乙酰基)-6-甲基吲哚類衍生物的用圖
- 一種黑霉菌生產(s)-乙酰基噻吩的方法
- 用于治療皮膚病的甲酰基肽受體2的激動劑的用圖
- 1-(1’,3’,4’,6’-四-o-乙酰基-d-葡萄糖)-4-對位取代芳基-[1,2,3]-三唑及制法和應用
- 一種從紅豆杉枝葉中分離提純10-去乙酰基巴卡亭iii的方法
- 一種從紅豆杉中提取分離10-去乙酰基巴卡丁ⅲ(10-dab ⅲ)的方法
- 乙酰基澳澤蘭素在制備抗腫瘤藥物中的應用
- 包含艾地苯醌、n-乙酰基-s-法呢基-l-半胱氨酸和麥角硫因的制劑及其用圖