載維生素A及其衍生物的環糊精?金屬有機骨架復合物和維生素A及其衍生物的深加工方法與流程
本發明屬于復合材料領域,尤其涉及一種載維生素A及其衍生物的環糊精-金屬有機骨架復合物和維生素A及其衍生物的深加工方法。
背景技術:
維生素A(Vitamin A)是一種重要、極易缺乏的,為人體維持正常代謝和機能所必需的脂溶性維生素,最早由美國科學家Elmer McCollum和Margaret Davis于1912-1914年之間發現,Margaret Davis等人從鱘魚肝臟中提取出一種黃色黏稠液體-維生素A。狹義維生素A即視黃醇,廣義維生素A還包括視黃醛、視黃酸、視黃醇乙酸酯和視黃醇棕櫚酸酯等在內的視黃醇衍生物,目前市售的維生素A主要為維生素A醋酸酯和維生素A棕櫚酸酯。維生素A具有多方面的生理功能,在維持視力、免疫功能、生長發育、抑制腫瘤生長、改善貧血等方面發揮著重要的作用。維生素A棕櫚酸酯(Vitamin A palmitate,VAP)與維生素A具有相似的結構,不溶于水,溶于乙醇,極易溶于正己烷、二氯甲烷、石油醚等非極性的有機溶劑,對酸、堿、光、熱、氧氣以及濕度敏感。維生素A及其衍生物作為人體必須的營養物質,機體自身不能合成,只能靠從食物中補充。為了保證機體正常的生理功能,必須提高維生素A及其衍生物的穩定性。
目前關于改善維生素A穩定性的液體制劑專利報道主要有維生素A納米乳(CN 103520101 B)、維生素A脂質體(CN 1245153C和CN105496801 A)和維生素A膠束(CN 103565676 B),這些液體制劑含有乳化劑和防腐劑,且所得的產品呈液體狀態,不利于包裝、存儲和運輸。改善維生素A穩定性的固體制劑的技術主要是微囊化,國內外關于維生素A微囊的專利報道主要有CN 10219816 A、CN 1022579397 B、US6124274、US6328995、US7279180和US20130177619等,但維生素A微囊需要在添加抗氧劑和表面活性劑的情況下才能使制劑中維生素A具有較好的穩定性。
技術實現要素:
有鑒于此,本發明的目的在于提供一種載維生素A及其衍生物的環糊精-金屬有機骨架復合物和維生素A及其衍生物的深加工方法,維生素A及其衍生物在本發明提供的復合物中具有較高的穩定性。
本發明提供了一種載維生素A及其衍生物的環糊精-金屬有機骨架復合物,包括:
環糊精-金屬有機骨架;
負載在所述骨架上的維生素,所述維生素為維生素A和/或其衍生物。
優選的,所述環糊精-金屬有機骨架包括經酸化的氫氧化鉀環糊精-金屬有機骨架材料、經酸化的碳酸鉀環糊精-金屬有機骨架材料、經交聯的氫氧化鉀環糊精-金屬有機骨架材料、經交聯的碳酸鉀環糊精-金屬有機骨架材料、酒石酸鉀環糊精-金屬有機骨架材料和醋酸鉀環糊精-金屬有機骨架材料中的一種或多種。
優選的,所述環糊精-金屬有機骨架的平均粒徑為50nm~50μm。
優選的,所述環糊精-金屬有機骨架的比表面積為500~1000m2/g。
優選的,所述復合物中維生素的含量≥2wt%。
優選的,所述維生素A衍生物包括視黃醛、視黃酸、視黃醇乙酸酯和視黃醇棕櫚酸酯中的一種或多種。
本發明提供了一種維生素A及其衍生物的深加工方法,包括以下步驟:
環糊精-金屬有機骨架和維生素在液體介質中混合,得到載維生素A及其衍生物的環糊精-金屬有機骨架復合物;
所述維生素為維生素A和/或其衍生物。
優選的,所述混合的方式為加熱攪拌。
優選的,所述加熱攪拌的溫度為20~60℃;所述加熱攪拌的轉數為200~800rpm;所述加熱攪拌的時間為0.5~6h。
優選的,所述環糊精-金屬有機骨架和維生素在液體介質中混合后,依次進行固液分離和干燥,得到載維生素A及其衍生物的環糊精-金屬有機骨架復合物。
與現有技術相比,本發明提供了一種載維生素A及其衍生物的環糊精-金屬有機骨架復合物和維生素A及其衍生物的深加工方法。本發明提供的復合物包括環糊精-金屬有機骨架;負載在所述骨架上的維生素,所述維生素為維生素A和/或其衍生物。本發明以環糊精-金屬有機骨架作為固體藥物儲庫,安全性高,生物相容性好;維生素填充在環糊精-金屬有機骨架中,具有較高的穩定性。實驗結果表明,本發明提供的復合物在60℃條件下熱處理10天后降解≤40%,40℃條件下放置90天后降解≤20%,熱穩定性優于巴斯夫維生素A粉。本發明提供的深加工方法包括以下步驟:環糊精-金屬有機骨架和維生素在液體介質中混合,得到載維生素A及其衍生物的環糊精-金屬有機骨架復合物;所述維生素為維生素A和/或其衍生物。本發明提供的深加工方法能夠制得穩定性良好的載維生素A及其衍生物的環糊精-金屬有機骨架復合物,該方法的工藝簡單可控,無需昂貴的設備。實驗結果表明,采用本發明提供的深加工方法制得的復合物載藥量高,晶型良好,粒徑均勻可控。
附圖說明
為了更清楚地說明本發明實施例或現有技術中的技術方案,下面將對實施例或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖僅僅是本發明的實施例,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據提供的附圖獲得其他的附圖。
圖1是本發明實施例1提供的酸化CD-MOFs-VAP復合物中維生素A棕櫚酸酯載藥量柱形圖;
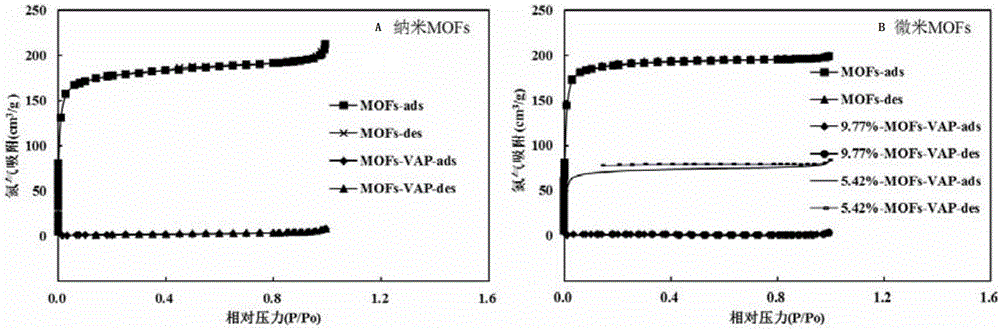
圖2是本發明實施例2提供的酸化CD-MOFs載藥前后的氣體吸附曲線圖;
圖3是本發明實施例3提供的酸化CD-MOFs-VAP復合物中維生素A棕櫚酸酯載藥量柱形圖;
圖4是本發明實施例4提供的SEM圖;
圖5是本發明實施例4提供的粒徑分布圖;
圖6是本發明實施例4提供的PXRD圖;
圖7是本發明實施例5提供的酸化CD-MOFs-VAP復合物中維生素A棕櫚酸酯載藥量柱形圖;
圖8是本發明實施例6提供的酸化CD-MOFs-VAP復合物中維生素A棕櫚酸酯載藥量柱形圖;
圖9是本發明實施例7提供的樣品于60℃條件下放置的降解曲線圖;
圖10是本發明實施例8提供的樣品于60℃條件下放置的降解曲線圖;
圖11是本發明實施例9提供的樣品于60℃條件下放置的降解曲線圖;
圖12是本發明實施例10提供的樣品于40℃條件下放置的降解曲線圖;
圖13是本發明實施例11提供的樣品于60℃條件下放置的降解曲線圖;
圖14是本發明實施例12提供的樣品于60℃條件下放置的降解曲線圖。
具體實施方式
下面對本發明實施例中的技術方案進行清楚、完整地描述,顯然,所描述的實施例僅僅是本發明一部分實施例,而不是全部的實施例。基于本發明中的實施例,本領域普通技術人員在沒有做出創造性勞動前提下所獲得的所有其他實施例,都屬于本發明保護的范圍。
本發明提供了一種載維生素A及其衍生物的環糊精-金屬有機骨架復合物,包括:
環糊精-金屬有機骨架;
復合在所述骨架上的維生素,所述維生素為維生素A和/或其衍生物。
本發明提供的復合物包括環糊精-金屬有機骨架和復合在所述骨架上的維生素。其中,所述環糊精-金屬有機骨架為環糊精與金屬鹽形成的骨架材料。在本發明中,所述環糊精包括但不限于:α-環糊精、β-環糊精、γ-環糊精、羥丙基-β-環糊精、磺丁基-β-環糊精、甲基-β-環糊精和羧甲基-β-環糊精中的一種或多少,優選γ-環糊精;所述金屬鹽中的金屬離子包括但不限于Li+、K+、Rb+、Cs+、Na+、Mg2+、Cd2+、Sn2+、Ag+、Yb+、Ba2+、Sr2+、Ca2+、Pb2+或La3+,優選為K+;所述金屬鹽中的陰離子包括但不限于OH-、NO3-、HCO3-、CO32-、C4H4O62-、CH3COO-、SCN-、C6H5COOH=C6H5COO-、Cl-、Br-、I-、O2-、S2-、HS-、HSO4-、ClO-、ClO3-或MnO4-,優選為OH-、CO32-、C4H4O62-或CH3COO-,更優選為OH-或CO32-。在本發明提供的一個實施例中,所述環糊精-金屬有機骨架包括但不限于經酸化的氫氧化鉀環糊精-金屬有機骨架材料、經酸化的碳酸鉀環糊精-金屬有機骨架材料、經交聯的氫氧化鉀環糊精-金屬有機骨架材料、經交聯的碳酸鉀環糊精-金屬有機骨架材料、酒石酸鉀環糊精-金屬有機骨架材料和醋酸鉀環糊精-金屬有機骨架材料中的一種或多種。在本發明中,所述環糊精-金屬有機骨架的平均粒徑優選為50nm~50μm,更優選為100nm~10μm,具體可為100nm~1000nm(即納米級)或1μm~10μm(即微米級);所述環糊精-金屬有機骨架的比表面積優選為500~1000m2/g,更優選為750~850m2/g。
本發明對所述環糊精-金屬有機骨架的來源沒有特別限定,按照本領域技術人員熟知的環糊精-金屬有機骨架制備方式進行制備即可,可以按照以下方式制備得到:
金屬鹽和環糊精在水中混合后,進行反應,得到環糊精-金屬有機骨架。
在本發明提供的上述制備方法中,直接將金屬鹽和環糊精在水中混合。其中,所述金屬鹽和環糊精的用量摩爾比優選(6~10):1為,更優選為8:1。混合后,加熱反應。其中,所述反應的溫度優選為40~70℃,更優選為50℃;所述反應的時間優選為1~4h,更優選為2h。反應結束后,靜置一段時間,得到環糊精-金屬有機骨架。在本發明中,為使所得環糊精-金屬有機骨架晶體能夠更快析出,優選在進行反應之前,在反應體系中預加一定量的有機溶劑;其中,所述有機溶劑包括但不限于甲醇、乙醇、異丙醇、丙酮和乙腈中的一種或多種;所述有機溶劑與環糊精的體積/質量比優選為(15~20)mL:1g,更優選為18.4mL:1g。在本發明中,為控制最終制得的環糊精-金屬有機骨架尺寸,優選在金屬鹽和環糊精反應一定時間后,在反應體系中加入尺寸調節劑;其中,所述尺寸調節劑包括但不限于聚乙二醇(簡稱:PEG)、聚乙二醇衍生物、聚維酮(簡稱:PVP)、聚維酮衍生物、失水山梨醇單月桂酸酯(簡稱:司盤)、失水山梨醇單月桂酸酯衍生物、聚氧乙烯月桂醇醚、聚氧乙烯月桂醇醚衍生物、乳化劑OP(其成分為壬烷基酚聚氧乙烯醚縮合物)、壬烷基酚聚氧乙烯醚縮合物衍生物、乳百靈A(其成分為聚氧乙烯脂肪醇醚)、聚氧乙烯脂肪醇醚衍生物、普流羅尼(其成分為聚氧乙烯聚丙二醇縮合物)、聚氧乙烯聚丙二醇縮合物衍生物、十二烷基硫酸鈉、十二烷基硫酸鈉衍生物、十二烷基苯磺酸鈉、十二烷基苯磺酸鈉衍生物、十六烷基三甲基溴化銨(簡稱:CTAB)、十六烷基三甲基溴化銨衍生物、十二烷基二甲基芐基溴化銨(簡稱:苯扎溴銨)和十二烷基二甲基芐基溴化銨衍生物中的一種或幾種;所述聚乙二醇優選為PEG 200、PEG 400、PEG 600、PEG 800、PEG 1000、PEG 1500、PEG 2000、PEG 4000、PEG 6000、PEG 8000、PEG 10000或PEG 20000,更優選為PEG 2000、PEG 4000、PEG 6000、PEG 8000、PEG 10000或PEG 20000,最優選為PEG 20000;所述聚維酮優選為PVP K12、PVP K15、PVP K17、PVP K25、PVP K30、PVP K60、PVP K90或PVP K120;所述失水山梨醇單月桂酸酯優選為司盤20、司盤40、司盤60或司盤80。所述尺寸調節劑與環糊精的質量比優選為2:(3~8),更優選為2:5。在本發明中,為提高制取的環糊精-金屬有機骨架的純凈度,優選對制得的環糊精-金屬有機骨架進行洗滌和干燥。
在本發明提供的一個實施例中,若環糊精-金屬有機骨架為經酸化的環糊精-金屬有機骨架,則可按照以下方式進行酸化處理:
將未酸化的環糊精-金屬有機骨架混懸于含酸的有機溶劑中,進行振搖孵育,得到經酸化的環糊精-金屬有機骨架。其中,所述酸包括但不限于冰醋酸、甲酸、檸檬酸、富馬酸、酒石酸、偏酒石酸、蘋果酸或己二酸,鹽酸、磷酸和硫酸中的一種或多種,優選冰醋酸;所述有機溶劑包括但不限于甲醇、乙醇、異丙醇、丙酮和乙腈中的一種或多種,優選乙醇;所述酸與有機溶劑的體積比優選為1:(5~20),更優選為1:10;所述振搖孵育的時間優選為0.5~6h,更優選為2h;所述振搖孵育的溫度優選為20~45℃,更優選為25℃。在本發明中,為提高制取的經酸化的環糊精-金屬有機骨架的純凈度,優選對制得的經酸化的環糊精-金屬有機骨架進行洗滌和干燥。
在本發明提供的一個實施例中,若環糊精-金屬有機骨架為經交聯的環糊精-金屬有機骨架,則可按照以下方式進行交聯處理:
在交聯劑與催化劑存在下,未交聯的環糊精-金屬有機骨架在介質中進行反應,得到經交聯的環糊精-金屬有機骨架。其中,所述反應介質包括但不限于二甲基甲酰胺、二甲基亞砜、二甲基乙酰胺、水或乙醇,優選為二甲基甲酰胺或二甲基亞砜,更優選為二甲基甲酰胺;所述交聯劑包括但不限于羰二咪唑、二碳酸酯、碳酸二苯酯、碳酸二甲酯、異氰酸酯、有機二酐或檸檬酸,優選為碳酸二苯酯;所述未交聯的環糊精-金屬有機骨架與交聯劑的質量比優選為(0.5~5):1,更優選為1:1;所述的催化劑包括但不限于N,N-二甲基環己胺、雙(2-二甲氨基乙基)醚、N,N,N',N'-四甲基亞烷基二胺、三乙胺、N,N-二甲基芐胺、固胺、N-乙基嗎啉、N,N’-二乙基哌嗪、三乙醇胺、N,N-二甲基乙醇胺(縮寫:DMEA)、吡啶或N,N’-二甲基吡啶,優選為三乙胺;所述未交聯的環糊精-金屬有機骨架與催化劑的質量/體積比優選為(1.5~2)g:1mL,更優選為1.7g:1mL;所述反應的溫度優選為60~90℃,更優選為70~80℃;所述反應的時間優選為1~48h,優選為4~24h,更優選為12~24h,最優選為24h。在本發明中,為提高制取的經交聯的環糊精-金屬有機骨架的純凈度,優選對制得的經交聯的環糊精-金屬有機骨架進行洗滌和干燥。
在本發明中,所述維生素負載在所述環糊精-金屬有機骨架上,即負載在所述環糊精-金屬有機骨架的表面和孔道中,所述維生素為維生素A和/或其衍生物。其中,所述維生素A又稱為視黃醇,所述維生素A衍生物包括但不限于視黃醛、視黃酸、視黃醇乙酸酯和視黃醇棕櫚酸酯中的一種或多種。在本發明中,所述復合中維生素的含量優選≥2wt%,更優選≥5wt%,最優選≥10wt%,具體可為5~15wt%;所述復合物中環糊精與維生素的摩爾比優選為(2~20):1,更優選為(3~8):1,最優選為(4~5):1。
在本發明中,60℃條件下熱處理10天,所述復合物中維生素的降解優選≤40%,更優選≤30%,最優選≤20%;40℃條件放置90天,所述復合物中維生素A及其衍生物的降解優選≤20%,更優選≤10%。
本發明以環糊精-金屬有機骨架作為藥物儲庫,安全性高,生物相容性好;維生素填充在環糊精-金屬有機骨架中,具有較高的穩定性。實驗結果表明,本發明提供的復合物在60℃條件下熱處理10天后降解≤40%,40℃條件下放置90天后降解≤20%,熱穩定性優于巴斯夫維生素A粉。
本發明提供了一種維生素A及其衍生物的加工方法,包括以下步驟:
環糊精-金屬有機骨架和維生素在液體介質中混合,得到載維生素A及其衍生物的環糊精-金屬有機骨架復合物;
所述維生素為維生素A和/或其衍生物。
在本發明提供的方法中,直接將環糊精-金屬有機骨架和維生素在液體介質中混合。其中,所述環糊精-金屬有機骨架和維生素在上文中已經介紹,在此不再贅述;所述液體介質包括但不限于壬醇(n-hexane)、二氯甲烷(DCM)、異丙醇(Isopropanol)、乙醇(Ethanol)、丙酮(acetone)、乙腈(acetonitrile)和二甲基甲酰胺(DMF)中的一種或多種;所述環糊精-金屬有機骨架和維生素的質量比優選為(1~10):(1~2),更優選為5:2。在本發明中,優選先將維生素與液體介質混合,得到混合液;再將環糊精-金屬有機骨架與所述混合液混合。其中,所述混合液中維生素的濃度優選為20~60mg/mL,具體可為30mg/mL、40mg/mL或50mg/mL。在本發明中,所述環糊精-金屬有機骨架和維生素在液體介質中進行混合的方式優選為加熱攪拌。其中,所述加熱攪拌的溫度優選為20~60℃,更優選為30~50℃,最優選為40℃;所述加熱攪拌的轉數優選為200~800rpm,更優選為300~600rpm,最優選為400rpm;所述加熱攪拌的時間優選為0.5~6h,更優選為1~4h,最優選為2h。
環糊精-金屬有機骨架和維生素在液體介質中混合一定時間后,得到載維生素A及其衍生物的環糊精-金屬有機骨架復合物。為將所述復合物從液體介質中分離出來,對優選對混合體系依次進行固液分離和干燥。其中,所述固液分離的方式優選為離心分離;所述離心分離的轉數優選為1000~5000rpm,更優選為2000~4000rpm,最優選為3000rpm;所述離心分離的時間優選為3~10min,更優選為4~8min,最優選為5min。所述干燥的方式優選為真空干燥;所述干燥的溫度優選為30~60℃,更優選為35~50℃,最優選為40℃。
本發明提供的加工方法能夠制得穩定性良好的載維生素A及其衍生物的環糊精-金屬有機骨架復合物,該方法的工藝簡單可控,無需昂貴的設備。實驗結果表明,采用本發明提供的加工方法制得的復合物載藥量高,晶型良好,粒徑均勻可控。
本發明提供的上述整體技術方案和優選技術方案,至少具有如下優點:
(a)本發明提供的復合物能顯著地改善維生素A及其衍生物的穩定性。
(b)本發明的提供的加工方法簡便快速,無需大型設備,在加工過程中無苛刻條件,節約了能源。
(c)本發明的提供的環糊精-金屬有機骨架方法制得的環糊精-金屬有機骨架材料的尺寸規則,產率高。
(d)本發明以環糊精作為主要原材料,對人體無害,利于工業化生產制備。
(e)在本發明提供的加工方法中,在環糊精-金屬有機骨架和維生素A及其衍生物進行混合時所用的液體介質優選為乙醇,無毒,對人體無害,適用于工業化生產。
(f)在本發明提供的加工方法中,環糊精-金屬有機骨架和維生素在液體介質中進行混合的方式優選為加熱攪拌,該方式非常簡便,易于實現。
為更清楚起見,下面通過以下實施例進行詳細說明。
在本申請的下述實施例中,為精簡描述,將γ-環糊精縮寫為γ-CD;將的環糊精-金屬有機骨架縮寫為CD-MOFs;氫氧化鉀環糊精-金屬有機骨架材料縮寫為KOH-CD-MOFs;將碳酸鉀環糊精-金屬有機骨架材料縮寫為K2CO3-CD-MOFs;將醋酸鉀環糊精-金屬有機骨架材料縮寫為CH3COOK-CD-MOFs;將載維生素A及其衍生物的環糊精-金屬有機骨架復合物縮寫為CD-MOFs-VAP復合物,其中VAP表示維生素A及其衍生物;
實施例1
不同粒徑的酸化CD-MOFs-VAP復合物的制備
將3.26gγ-CD、1.12g KOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級CD-MOFs(平均粒徑1~10μm),pH約為11~13,產率約80%。
將3.26gγ-CD、1.12g KOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱2h。反應一定時間后,加入等體積的甲醇后,再加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性納米級CD-MOFs(平均粒徑100~1000nm),pH約為11~13,產率約80%。
分別稱取一定量上述制備的堿性微米級CD-MOFs和堿性納米級CD-MOFs,分散于含有冰醋酸的無水乙醇中(冰醋酸與無水乙醇的體積比為1:10),置于搖床中在25℃下振搖孵育2h后,用乙醇洗滌,收集固體,真空干燥,即得經酸化的微米級CD-MOFs和經酸化的納米級CD-MOFs。若將所述經酸化的CD-MOFs的溶解于水中,制成10mg/mL的水溶液,其pH約為5-8。
分別稱取上述制備的經酸化的微米級CD-MOFs和經酸化的納米級CD-MOFs,加入一定量維生素A棕櫚酸酯濃度為40mg/mL的乙醇溶液,使CD-MOFs與維生素A棕櫚酸酯的投料質量比為5:2,40℃下加熱攪拌2h后,離心(3000rpm,5min),下層沉淀物分為兩份,一份用乙醇洗滌,一份不洗滌,兩份下沉物均在40℃下真空干燥,即得洗滌的載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物、未洗滌的載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物,以及洗滌的載維生素A棕櫚酸酯的酸化納米級CD-MOFs復合物、未洗滌的載維生素A棕櫚酸酯的酸化納米級CD-MOFs復合物。
復合物中維生素A棕櫚酸酯含量測試:
稱取一定量上述載維生素A棕櫚酸酯的酸化CD-MOFs復合物,溶于一定量的水和異丙醇中,高效液相色譜法測定維生素A棕櫚酸酯含量。計算樣品中維生素A棕櫚酸酯的含量(即載藥量),維生素A棕櫚酸酯含量=樣品中維生素A棕櫚酸酯的重量/樣品總重量×100%。
維生素A棕櫚酸酯載藥量測試結果如圖1所示,圖1是本發明實施例1提供的酸化CD-MOFs-VAP復合物中維生素A棕櫚酸酯載藥量柱形圖。通過圖1可以看出,酸化微米級CD-MOFs空腔內容納的維生素A棕櫚酸酯比酸化納米級CD-MOFs多,但酸化納米級CD-MOFs表面吸附的維生素A棕櫚酸酯比酸化微米級CD-MOFs多。
實施例2
不同粒徑的酸化CD-MOFs-VAP復合物的比表面積考察
按照實施例1的方法,調整CD-MOFs與維生素A棕櫚酸酯的投料質量比,分別制備:酸化納米級CD-MOFs、未洗滌的酸化納米級CD-MOFs-VAP復合物(VAP含量為14.11wt%)、酸化微米級CD-MOFs、未洗滌的酸化微米級CD-MOFs-VAP復合物(VAP含量為9.77wt%)和未洗滌的酸化微米級CD-MOFs-VAP復合物(VAP含量為5.42wt%)。
分別對上述六種樣品進行氣體吸附測試,具體測試過程為:稱取100-200mg待測樣品,氣體吸附前對樣品在50℃下脫氣4-5h,液氮溫度(77K)下測定CD-MOF對N2吸附效果。氣體吸附結果如圖2所示,圖2是本發明實施例2提供的酸化CD-MOFs載藥前后的氣體吸附曲線圖,其中圖A為納米級CD-MOFs氣體吸附曲線,圖B為微米級CD-MOFs氣體吸附曲線,圖中-ads表示樣品吸附N2過程,-des表示樣品解吸附過程。通過圖2可以看出,酸化納米級CD-MOFs的比表面積為762.45m2/g,酸化納米級CD-MOFs-VAP復合物(VAP含量為14.11wt%)的比表面積為9.58m2/g;酸化微米級CD-MOFs的比表面積為823.12m2/g,酸化微米級CD-MOFs-VAP復合物(VAP含量為9.77wt%)的比表面積為3.84m2/g,酸化微米級CD-MOFs-VAP復合物(VAP含量為5.42wt%)的比表面積為314.03m2/g。可見,本發明實施例提供的CD-MOFs-VAP復合物具有較高的載藥量(載藥量越高比表面積降低的越顯著)。
實施例3
在不同投藥濃度下制備酸化微米級CD-MOFs-VAP復合物
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級CD-MOFs(平均粒徑1~10μm),產率約80%。
稱取一定量上述制備的堿性微米級CD-MOFs,分散于含有冰醋酸的無水乙醇中(冰醋酸與無水乙醇的體積比為1:10),置于搖床中在25℃下振搖孵育2h后,用乙醇洗滌,收集固體,真空干燥,即得經酸化的微米級CD-MOFs。
稱取三份上述制備的酸化微米級CD-MOFs,每份200mg,依次加入2mL濃度分別為30、40、50mg/mL的維生素A棕櫚酸酯的乙醇溶液,40℃下加熱攪拌2h后,離心(3000rpm,5min),下層沉淀物分為兩份,一份用乙醇洗滌,一份不洗滌,兩份下沉物均在40℃下真空干燥,即得洗滌的載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物、未洗滌的載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物。
復合物中維生素A棕櫚酸酯含量測試:
稱取一定量上述載維生素A棕櫚酸酯的酸化微米級CD-MOF復合物,溶于一定量的水和異丙醇中,高效液相色譜法測定維生素A棕櫚酸酯含量。計算樣品中維生素A棕櫚酸酯的含量(即載藥量),維生素A棕櫚酸酯含量=樣品中維生素A棕櫚酸酯的重量/樣品總重量×100%。
維生素A棕櫚酸酯載藥量測試結果如圖3所示,圖3是本發明實施例3提供的酸化CD-MOFs-VAP復合物中維生素A棕櫚酸酯載藥量柱形圖。通過圖3可以看出,無論洗滌還是未洗滌的載維生素A棕櫚酸酯的酸化微米級CD-MOF復合物,投藥濃度為40mg/mL時,載藥量達到飽和。
實施例4
酸化微米級CD-MOFs-VAP復合物的粒徑和晶體形態考察
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級CD-MOFs(平均粒徑1~10μm),產率約80%。
稱取一定量上述制備的堿性微米級CD-MOFs,分散于含有冰醋酸的無水乙醇中(冰醋酸與無水乙醇的體積比為1:10),置于搖床中在25℃下振搖孵育2h后,用乙醇洗滌,收集固體,真空干燥,即得經酸化的微米級CD-MOFs。
稱取上述制備的經酸化的微米級CD-MOFs,加入一定量維生素A棕櫚酸酯濃度為40mg/mL的乙醇溶液,使CD-MOFs與維生素A棕櫚酸酯的投料質量比為5:2,40℃下加熱攪拌2h后,離心(3000rpm,5min),下層沉淀物在40℃下真空干燥,即得載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物。
分別對上述制備的堿性微米級CD-MOFs、經酸化的微米級CD-MOFs和載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物進行掃描電子顯微鏡(SEM)測試、粒徑分布測試和多晶X射線衍射(PXRD)測試。結果如圖4、圖5和圖6所示,圖4是本發明實施例4提供的SEM圖,圖4中A為堿性微米級CD-MOFs、B為經酸化的微米級CD-MOFs、C為載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物;圖5是本發明實施例4提供的粒徑分布圖,圖5中A為堿性微米級CD-MOFs、B為經酸化的微米級CD-MOFs、C為載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物;圖6是本發明實施例4提供的PXRD圖,圖6中A為堿性微米級CD-MOFs、B為經酸化的微米級CD-MOFs、C為載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物。通過圖4、5、6可以看出,將堿性微米級CD-MOFs酸化和載藥后,CD-MOFs的結晶度及晶體形態沒有改變,粒徑(2~3μm)較均一。
實施例5
在不同載藥溶劑中制備酸化微米級CD-MOFs-VAP復合物
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級CD-MOFs(平均粒徑1~10μm),產率約80%。
稱取一定量上述制備的堿性微米級CD-MOFs,分散于含有冰醋酸的無水乙醇中(冰醋酸與無水乙醇的體積比為1:10),置于搖床中在25℃下振搖孵育2h后,用乙醇洗滌,收集固體,真空干燥,即得經酸化的微米級CD-MOFs。
稱取上述制備的經酸化的微米級CD-MOFs,分別加入含80mg維生素A棕櫚酸酯的正己烷2mL、二氯甲烷2mL、異丙醇2mL、乙醇2mL、丙酮2mL、乙腈40mL、N,N二甲基甲酰胺2mL,使CD-MOFs與維生素A棕櫚酸酯的投料質量比為5:2,40℃下加熱攪拌2h后,離心(3000rpm,5min),下層沉淀物在40℃下真空干燥,即得載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物。
稱取一定量上述載維生素A棕櫚酸酯的酸化微米級CD-MOF復合物,溶于一定量的水和異丙醇中,高效液相色譜法測定維生素A棕櫚酸酯含量。計算樣品中維生素A棕櫚酸酯的含量(即載藥量),維生素A棕櫚酸酯含量=樣品中維生素A棕櫚酸酯的重量/樣品總重量×100%。
維生素A棕櫚酸酯載藥量測試結果如圖7所示,圖7是本發明實施例5提供的酸化CD-MOFs-VAP復合物中維生素A棕櫚酸酯載藥量柱形圖,圖7中,A為單一溶劑圖,B為混合溶劑圖。通過圖7可以看出,乙醇是最佳的載藥溶劑。
實施例6
在不同加熱攪拌條件下制備酸化微米級CD-MOFs-VAP復合物
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級CD-MOFs(平均粒徑1~10μm),產率約80%。
稱取一定量上述制備的堿性微米級CD-MOFs,分散于含有冰醋酸的無水乙醇中(冰醋酸與無水乙醇的體積比為1:10),置于搖床中在25℃下振搖孵育2h后,用乙醇洗滌,收集固體,真空干燥,即得經酸化的微米級CD-MOFs。
稱取上述制備的經酸化的微米級CD-MOFs,加入一定量維生素A棕櫚酸酯濃度為40mg/mL的乙醇溶液,使CD-MOFs與維生素A棕櫚酸酯的投料質量比為5:2,分別在30、40、50℃下加熱攪拌1、2、4h后,離心(3000rpm,5min),下層沉淀物在40℃下真空干燥,即得載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物。
稱取一定量上述載維生素A棕櫚酸酯的酸化微米級CD-MOF復合物,溶于一定量的水和異丙醇中,高效液相色譜法測定維生素A棕櫚酸酯含量。計算樣品中維生素A棕櫚酸酯的含量(即載藥量),維生素A棕櫚酸酯含量=樣品中維生素A棕櫚酸酯的重量/樣品總重量×100%。
維生素A棕櫚酸酯載藥量測試結果如圖8所示,圖8是本發明實施例6提供的酸化CD-MOFs-VAP復合物中維生素A棕櫚酸酯載藥量柱形圖,通過圖8可以看出,加熱攪拌的溫度和時間對載藥量有一定影響,最佳的載藥溫度和時間分別為40℃和2h。
實施例7
洗滌/未洗滌的酸化微米級CD-MOFs-VAP復合物的熱穩定性考察
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級CD-MOFs(平均粒徑1~10μm),產率約80%。
稱取一定量上述制備的堿性微米級CD-MOFs,分散于含有冰醋酸的無水乙醇中(冰醋酸與無水乙醇的體積比為1:10),置于搖床中在25℃下振搖孵育2h后,用乙醇洗滌,收集固體,真空干燥,即得經酸化的微米級CD-MOFs。
稱取上述制備的經酸化的微米級CD-MOFs,加入一定量維生素A棕櫚酸酯濃度為40mg/mL的乙醇溶液,使CD-MOFs與維生素A棕櫚酸酯的投料質量比為5:2,40℃下加熱攪拌2h后,離心(3000rpm,5min),下層沉淀物分為兩份,一份用乙醇洗滌,一份不洗滌,兩份下沉物均在40℃下真空干燥,即得洗滌的載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物和未洗滌的載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物。
稱取一定量上述載維生素A棕櫚酸酯的酸化微米級CD-MOF復合物,以及物理混合物(分別稱取酸化微米級CD-MOFs和維生素A棕櫚酸酯置于研缽中,研磨均勻,即得物理混合物,CD-MOFs與維生素A棕櫚酸酯的質量比為5:4)于西林瓶中,封口置于60℃烘箱中,于0、5、10d時,分別精密稱取一定量的樣品,溶于一定量的水和異丙醇中,高效液相色譜法測定維生素A棕櫚酸酯含量。維生素A棕櫚酸酯的含量計算方法同實施例1,結果為:
1)、0d時,洗滌的載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物中維生素A棕櫚酸酯的含量(w/w)為6%,其中環糊精與維生素A棕櫚酸酯的摩爾比為6.3:1(環糊精與維生素A棕櫚酸酯摩爾比=(復合物中CD-MOFs的質量/環糊精的摩爾質量)/(復合物中維生素A棕櫚酸酯的質量/維生素A棕櫚酸酯的摩爾質量));未洗滌的載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物中維生素A棕櫚酸酯的含量(w/w)為7%,其中環糊精與維生素A棕櫚酸酯的摩爾比為5.0:1;物理混合中維生素A棕櫚酸酯的含量(w/w)為8%。
2)、0、5、10d時,熱穩定性變化如圖9所示,圖9是本發明實施例7提供的樣品于60℃條件下放置的降解曲線圖,通過圖9可以看出,洗滌的載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物中維生素A棕櫚酸酯60℃下10天后降解45%;未洗滌的載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物中維生素A棕櫚酸酯10天后降解38%;物理混合物中維生素A棕櫚酸酯10天后降解59%。洗滌與未洗滌的復合物與其物理混合物相比,維生素A棕櫚酸酯的熱穩定性明顯提高,且未洗滌的復合物對維生素A棕櫚酸酯的熱穩定性保護作用優于洗滌的復合物。
實施例8
不同粒徑的酸化CD-MOFs-VAP復合物的熱穩定性考察
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級CD-MOFs(平均粒徑1~10μm),產率約80%。
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入等體積的甲醇后,再加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性納米級CD-MOFs(平均粒徑100~1000nm),產率約80%。
分別稱取一定量上述制備的堿性微米級CD-MOFs和堿性納米級CD-MOFs,分散于含有冰醋酸的無水乙醇中(冰醋酸與無水乙醇的體積比為1:10),置于搖床中在25℃下振搖孵育2h后,用乙醇洗滌,收集固體,真空干燥,即得經酸化的微米級CD-MOFs和經酸化的納米級CD-MOFs。
分別稱取上述制備的經酸化的微米級CD-MOFs和經酸化的納米級CD-MOFs,加入一定量維生素A棕櫚酸酯濃度為40mg/mL的乙醇溶液,使CD-MOFs與維生素A棕櫚酸酯的投料質量比為5:2,40℃下加熱攪拌2h后,離心(3000rpm,5min),下層沉淀物在40℃下真空干燥,即得載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物和載維生素A棕櫚酸酯的酸化納米級CD-MOFs復合物。
稱取一定量上述載維生素A棕櫚酸酯的酸化微米級CD-MOF復合物,以及巴斯夫維生素A粉于西林瓶中,封口置于60℃烘箱中,于0、5、10d時,分別精密稱取一定量的樣品,溶于一定量的水和異丙醇中,高效液相色譜法測定維生素A棕櫚酸酯含量。維生素A棕櫚酸酯的含量計算方法同實施例1,結果為:
1)0d時,載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物中維生素A棕櫚酸酯的含量(w/w)為10%,其中環糊精與維生素A棕櫚酸酯的摩爾比為3.6:1;載維生素A棕櫚酸酯的酸化納米級CD-MOFs復合物中維生素A棕櫚酸酯的含量(w/w)為11%,其中環糊精與維生素A棕櫚酸酯的摩爾比為3.3:1;巴斯夫維生素A粉中維生素A醋酸酯的含量(w/w)為10%。
2)0、5、10d時,熱穩定性變化如圖10所示,圖10是本發明實施例8提供的樣品于60℃條件下放置的降解曲線圖,通過圖10可以看出,載維生素A棕櫚酸酯的酸化納米級CD-MOFs復合物中維生素A棕櫚酸酯60℃下10天后降解76%,載維生素A棕櫚酸酯的酸化微米級CD-MOFs復合物中維生素A棕櫚酸酯10天后降解29%,巴斯夫維生素A粉中維生素A醋酸酯10天后降解41%。酸化微米級CD-MOFs對維生素A的保護作用優于酸化納米級CD-MOFs和巴斯夫維生素A粉。
實施例9
不同金屬鹽的酸化CD-MOFs-VAP復合物的短期熱穩定性考察
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級KOH-CD-MOFs(平均粒徑1~10μm),產率約80%。
將3.26gγ-CD、1.38gK2CO3溶于100mL水中,預加適量100mL甲醇至γ-CD與K2CO3混合溶液內,密閉,60℃加熱處理2h。反應一定時間后,加入1.6g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級K2CO3-CD-MOFs(平均粒徑1~10μm),產率約50%。
分別稱取一定量上述制備的堿性微米級KOH-CD-MOFs和堿性微米級K2CO3-CD-MOFs,分散于含有冰醋酸的無水乙醇中(冰醋酸與無水乙醇的體積比為1:10),置于搖床中在25℃下振搖孵育2h后,用乙醇洗滌,收集固體,真空干燥,即得經酸化的微米級KOH-CD-MOFs和經酸化的微米級K2CO3-CD-MOFs。
分別稱取上述制備的經酸化的微米級KOH-CD-MOFs和經酸化的微米級K2CO3-CD-MOFs,加入一定量維生素A棕櫚酸酯濃度為40mg/mL的乙醇溶液,使CD-MOFs與維生素A棕櫚酸酯的投料質量比為5:2,40℃下加熱攪拌2h后,離心(3000rpm,5min),下層沉淀物分為兩份,一份用乙醇洗滌,一份不洗滌,兩份下沉物均在40℃下真空干燥,即得洗滌的載維生素A棕櫚酸酯的酸化微米級KOH-CD-MOFs復合物、未洗滌的載維生素A棕櫚酸酯的酸化微米級KOH-CD-MOFs復合物,以及洗滌的載維生素A棕櫚酸酯的酸化微米級K2CO3-CD-MOFs復合物、未洗滌的載維生素A棕櫚酸酯的酸化微米級K2CO3-CD-MOFs復合物。
稱取一定量上述載維生素A棕櫚酸酯的酸化微米級CD-MOF復合物,以及巴斯夫維生素A粉于西林瓶中,封口置于60℃烘箱中,于0、1、2、3、4、7、9d時,分別精密稱取一定量的樣品,溶于一定量的水和異丙醇中,高效液相色譜法測定維生素A棕櫚酸酯含量,維生素A棕櫚酸酯的含量計算方法同實施例1,之后計算降解,降解結果如圖11所示,圖11是本發明實施例9提供的樣品于60℃條件下放置的降解曲線圖。通過圖11可以看出,洗滌的載維生素A棕櫚酸酯的酸化微米級KOH-CD-MOFs復合物9天后分別降解50%,未洗滌的載維生素A棕櫚酸酯的酸化微米級KOH-CD-MOFs復合物9天后分別降解49%,洗滌的載維生素A棕櫚酸酯的酸化微米級K2CO3-CD-MOFs復合物9天后分別降解43%,未洗滌的載維生素A棕櫚酸酯的酸化微米級K2CO3-CD-MOFs復合物9天后分別降解32%,巴斯夫維生素A粉中維生素A9天后降解53%。短期穩定性考察中性微米級CD-MOFs-VAP穩定性優于巴斯夫維生素A粉。
實施例10
不同金屬鹽的酸化CD-MOFs-VAP復合物的長期熱穩定性考察
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加適量60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級KOH-CD-MOFs(平均粒徑1~10μm),產率約80%。
將3.26gγ-CD、1.38gK2CO3溶于100mL水中,預加100mL甲醇至γ-CD與K2CO3混合溶液內,密閉,60℃加熱2h。反應一定時間后,加入1.6g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級K2CO3-CD-MOFs(平均粒徑1~10μm),產率約50%。
分別稱取一定量上述制備的堿性微米級KOH-CD-MOFs和堿性微米級K2CO3-CD-MOFs,分散于含有冰醋酸的無水乙醇中(冰醋酸與無水乙醇的體積比為1:10),置于搖床中在25℃下振搖孵育2h后,用乙醇洗滌,收集固體,真空干燥,即得經酸化的微米級KOH-CD-MOFs和經酸化的微米級K2CO3-CD-MOFs。
分別稱取上述制備的經酸化的微米級KOH-CD-MOFs和經酸化的微米級K2CO3-CD-MOFs,加入一定量維生素A棕櫚酸酯濃度為40mg/mL的乙醇溶液,使CD-MOFs與維生素A棕櫚酸酯的投料質量比為5:2,40℃下加熱攪拌2h后,離心(3000rpm,5min),下層沉淀物在40℃下真空干燥,即得載維生素A棕櫚酸酯的酸化微米級KOH-CD-MOFs復合物和載維生素A棕櫚酸酯的酸化微米級K2CO3-CD-MOFs復合物。
稱取一定量上述載維生素A棕櫚酸酯的酸化微米級CD-MOF復合物、物理混合物(分別稱取酸化微米級CD-MOFs和維生素A棕櫚酸酯置于研缽中,研磨均勻,即得物理混合物,CD-MOFs與維生素A棕櫚酸酯的質量比為19:1)以及巴斯夫維生素A粉于西林瓶中,封口置于40℃烘箱中,于0、1、2、3月時,分別精密稱取一定量的樣品,溶于一定量的水和異丙醇中,高效液相色譜法測定維生素A棕櫚酸酯含量,維生素A棕櫚酸酯的含量計算方法同實施例1,之后計算降解,降解結果如圖12所示,圖12是本發明實施例10提供的樣品于40℃條件下放置的降解曲線圖。通過圖12可以看出,載維生素A棕櫚酸酯的酸化微米級KOH-CD-MOFs復合物中維生素A棕櫚酸酯3月后降解11%,載維生素A棕櫚酸酯的酸化微米級K2CO3-CD-MOFs復合物中維生素A棕櫚酸酯3月后降解7%,巴斯夫維生素A粉中維生素A 3月后降解后降解12%,物理混合物3月后降解42%。
實施例11
交聯CD-MOFs-VAP復合物的制備和熱穩定性考察
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級CD-MOFs(平均粒徑1~10μm),產率約80%。
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入等體積的甲醇后,再加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性納米級CD-MOFs(平均粒徑100~1000nm),產率約80%。
分別稱取0.778g上述堿性微米級CD-MOFs或堿性納米級CD-MOF,加入10mL的DMF后,加入0.771g的碳酸二苯酯和0.45mL三乙胺,80℃攪拌24h,倒入乙醇中析出,離心后,水洗1次,丙酮洗滌2次,真空干燥,得交聯的微米級CD-MOFs或交聯的納米級CD-MOF。
分別稱取上述制備的交聯的微米級CD-MOFs或交聯的納米級CD-MOF,加入一定量維生素A棕櫚酸酯濃度為40mg/mL的乙醇溶液,使CD-MOFs與維生素A棕櫚酸酯的投料質量比為5:2,40℃下加熱攪拌2h后,離心(3000rpm,5min),下層沉淀物在40℃下真空干燥,即得載維生素A棕櫚酸酯的交聯微米級CD-MOFs復合物或載維生素A棕櫚酸酯的交聯納米級CD-MOFs復合物。
稱取一定量上述載維生素A棕櫚酸酯的交聯微米級CD-MOFs復合物和載維生素A棕櫚酸酯的交聯納米級CD-MOFs復合物以及巴斯夫維生素A粉于西林瓶中,封口置于60℃烘箱中,于0、5、10d時,分別精密稱取一定量的樣品,溶于一定量的水和異丙醇中,高效液相色譜法測定維生素A棕櫚酸酯含量。維生素A棕櫚酸酯的含量計算方法同實施例1,結果為:
1)0d時,載維生素A棕櫚酸酯的交聯微米級CD-MOFs復合物中維生素A棕櫚酸酯的含量(w/w)為8.2%,其中環糊精與維生素A棕櫚酸酯的摩爾比為4.5:1;載維生素A棕櫚酸酯的交聯納米級CD-MOFs復合物中維生素A棕櫚酸酯的含量(w/w)為7.7%,其中環糊精與維生素A棕櫚酸酯的摩爾比為4.9:1;巴斯夫維生素A粉中維生素A醋酸酯的含量(w/w)為10%。
2)0、5、10d時,熱穩定性變化如圖13所示,圖13是本發明實施例11提供的樣品于60℃條件下放置的降解曲線圖,通過圖13可以看出,載維生素A棕櫚酸酯的交聯納米級CD-MOFs復合物中維生素A棕櫚酸酯60℃下10天后降解39%,載維生素A棕櫚酸酯的交聯微米級CD-MOFs復合物中維生素A棕櫚酸酯10天后降解42%,巴斯夫維生素A粉中維生素A醋酸酯10天后降解41%。
實施例12
不同金屬鹽的CD-MOFs-VAP復合物的熱穩定性考察
將3.26gγ-CD、1.96gCH3COOK溶于50mL水中,預加30mL甲醇至γ-CD與CH3COOK混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入640mg聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得微米級CH3COOK-CD-MOFs(平均粒徑1~10μm),產率約50%。
將3.26gγ-CD、1.12gKOH溶于100mL水中,預加60mL甲醇至γ-CD與KOH混合溶液內,密閉,50℃加熱處理2h。反應一定時間后,加入1.28g聚乙二醇20000,室溫靜置一段時間后,用乙醇和二氯甲烷洗滌,將所得晶體真空干燥,即得堿性微米級KOH-CD-MOFs(平均粒徑1~10μm),產率約80%。
稱取一定量上述制備的堿性微米級KOH-CD-MOFs,分散于含有冰醋酸的無水乙醇中(冰醋酸與無水乙醇的體積比為1:10),置于搖床中在25℃下振搖孵育2h后,用乙醇洗滌,收集固體,真空干燥,即得經酸化的微米級KOH-CD-MOFs。
分別稱取上述制備的酸化微米級KOH-CD-MOFs和微米級CH3COOK-CD-MOFs,加入一定量維生素A棕櫚酸酯濃度為40mg/mL的乙醇溶液,使CD-MOFs與維生素A棕櫚酸酯的投料質量比為5:2,40℃下加熱攪拌2h后,離心(3000rpm,5min),下層沉淀物在40℃下真空干燥,即得載維生素A棕櫚酸酯的酸化微米級KOH-CD-MOFs復合物和載維生素A棕櫚酸酯的微米級CH3COOK-CD-MOFs復合物。
分別稱取一定量上述載維生素A棕櫚酸酯的酸化微米級KOH-CD-MOFs復合物和載維生素A棕櫚酸酯的微米級CH3COOK-CD-MOFs復合物于西林瓶中,封口置于60℃烘箱中,于0、5、10d時,分別精密稱取一定量的樣品,溶于一定量的水和異丙醇中,高效液相色譜法測定維生素A棕櫚酸酯含量。維生素A棕櫚酸酯的含量計算方法同實施例1,結果為:
1)0d時,載維生素A棕櫚酸酯的微米級CH3COOK-CD-MOFs復合物中維生素A棕櫚酸酯的含量(w/w)為5.3%,其中環糊精與維生素A棕櫚酸酯的摩爾比為7.2:1;載維生素A棕櫚酸酯的酸化微米級KOH-CD-MOFs復合物中維生素A棕櫚酸酯的含量(w/w)為5.7%,其中環糊精與維生素A棕櫚酸酯的摩爾比為6.7:1。
2)0、5、10d時,熱穩定性變化如圖14所示,圖14是本發明實施例12提供的樣品于60℃條件下放置的降解曲線圖,通過圖14可以看出,載維生素A棕櫚酸酯的酸化微米級KOH-CD-MOFs復合物中維生素A棕櫚酸酯60℃下10天后降解36%,載維生素A棕櫚酸酯的微米級CH3COOK-CD-MOFs復合物中維生素A棕櫚酸酯10天后降解56%。酸化微米級KOH-CD-MOFs對維生素A棕櫚酸酯的保護作用優于微米級CH3COOK-CD-MOFs。
以上所述僅是本發明的優選實施方式,應當指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!