一種磁分離RCA合成DNA酶檢測金黃色葡萄球菌的方法與流程
本發明涉及一種磁分離rca合成dna酶檢測金黃色葡萄球菌的方法,屬于食品安全技術領域。
背景技術:
金黃色葡萄球菌(staphylococcusaureus)大量存在于自然界中,是一種典型的球型革蘭氏陽性菌。金葡菌對生長環境要求不嚴,肉類、魚類、乳制品等絕大多數食品都可為其提供良好的生長條件,在適宜的溫度下,可產生耐高溫的腸毒素從而引發食物中毒。我國每年發生大量由金葡菌引起的食物中毒事件,其主要表現癥狀為嘔吐和腹瀉。因此,對金葡菌作出快速、精確的檢測十分重要。
傳統檢測金葡菌的方法為根據國標法對其進行分離培養和生化鑒別,存在檢測耗時、操作繁瑣、檢測限高等缺點,無法達到現今食品中微生物安全性的要求。隨著生物技術的快速發展,許多新型快速檢測技術得到了廣泛的研究和應用,如pcr技術、免疫學技術、生物傳感器和基因芯片技術等。
滾環擴增技術(rollingcircleamplification,rca)是近幾年發展起來的一種等溫條件下的dna復制技術,以短寡核苷酸鏈為引物,線性單鏈dna序列形成環狀模板,運用具有鏈置換作用的dna聚合酶使引物生成包含若干串聯重復序列互補環狀模板的線性長鏈dna分子。rca具有操作簡單、高特異性、擴增效率高、產物穩定等優點。dna酶是一段能形成g-四鏈體的核苷酸序列結合氯化血紅素后模擬過氧化物酶功能的復合物,可催化h2o2與魯米諾的化學發光反應。滾環擴增合成dna酶,結合了滾環擴增的信號放大作用和dna酶的催化作用,雙重放大。
近年來,許多研究將dna夾心雜交原理結合納米材料應用于致病菌的檢測,但其中也存在著一些檢測限高、靈敏度低和操作復雜等問題。因此,有必要建立一種檢測限低、靈敏度高且操作簡單的檢測金黃色葡萄球菌的方法。
技術實現要素:
為了解決目前存在的金黃色葡萄球菌檢測限高等問題,本發明提供了一種磁分離滾環擴增合成dna酶檢測金黃色葡萄球菌的方法。本發明方法,將金黃色葡萄球菌的16srdna為一段特異性檢測的目標dna片段,使用特異性探針修飾的磁納米粒子來結合和分離金葡菌的16srdna,不僅利用磁納米粒子的磁性分離作用,還利用探針和金葡菌16srdna間特異性的互補配對作用,從而快速地從復雜組分中分離金葡菌16srdna,并有效結合滾環擴增技術合成的dna酶催化h2o2-魯米諾反應系統來實現信號的雙重放大和檢測。
本發明利用dna夾心雜交原理設計了兩條特異性的探針——捕獲探針和信號探針。將捕獲探針修飾到磁納米粒子表面,該材料可快速地從復雜組分中結合和分離金黃色葡萄球菌的目標dna序列,再加入包含滾環擴增引物的信號探針與目標dna序列互補雜交形成夾心結構,然后滾環擴增引物與相應包含dna酶互補序列的模板互補配對引發滾環擴增反應合成dna酶,利用dna酶進行催化h2o2-魯米諾化學發光反應系統,根據相對發光值的變化來定量檢測金葡菌。在一定范圍內,金黃色葡萄球菌的濃度與相對發光值的變化呈線性相關,可達到對金黃色葡萄球菌定量檢測的目的。
本發明方法,包括如下步驟:
(5)先將捕獲探針與親和素修飾的磁納米粒子(avidin-mnps)混勻、孵育,使捕獲探針修飾到磁納米粒子表面,得到捕獲探針功能化的磁納米粒子;所述捕獲探針功能化的磁納米粒子能夠結合金黃色葡萄球菌的目標dna序列;
(6)將捕獲探針功能化的磁納米粒子與含目標dna序列的待測液孵育,然后加入信號探針,使目標dna序列與捕獲探針、信號探針生成夾心結構;所述信號探針上包含滾環擴增引物;
(7)加入滾環擴增模板,然后滾環擴增引物與相應包含dna酶互補序列的模板互補配對引發滾環擴增反應合成dna酶;
(8)利用dna酶進行催化h2o2-魯米諾化學發光反應系統,根據相對發光值的變化來定量檢測金黃色葡萄球菌。
在一種實施方式中,所述方法還包括先配制一定含有不同濃度的目標dna序列的標準樣品,建立不同濃度的目標dna序列與相對發光值的標準曲線,當檢測待測樣品時,將利用待測樣品得到的相對發光值代入到標準曲線中,即得到待測樣品中的目標dna序列的濃度。
在一種實施方式中,所述目標dna序列為cctttgacaactctagagatagagccttc(如seqidno:1所示)。
在一種實施方式中,所述金黃色葡萄球菌是來源于食品中的金黃色葡萄球菌。
在一種實施方式中,所述含目標dna序列的待測液,是提取食品中的金黃色葡萄球菌基因組得到的。
在一種實施方式中,所述捕獲探針的序列(如seqidno:2所示)為:biotin-ttttttgaaggctctatctct。
在一種實施方式中,所述信號探針的序列為(如seqidno:3所示):agagttgtcaaaggttttttaggatcgtgtggtt。
在一種實施方式中,所述dna酶的序列為:tggctgagttttgggttgggcgggttgggt(如seqidno:5所示)。
在一種實施方式中,所述相應包含dna酶互補序列的模板序列為(5’-3’):accgactcaaaacccaacccgccctacccaaaacccaacccgccctacccaaacatacacacagca(如seqidno:4所示)。
在一種實施方式中,所述親和素修飾的磁納米粒子的制備方法是:使用戊二醛交聯法,將氨基化的磁納米粒子加入到戊二醛和pbs溶液中,混合均勻后室溫下避光反應2h,用pbs緩沖液清洗3次后,再加入親和素溶液震蕩避光反應6h,pbs緩沖液清洗3次,即獲得親和素修飾的磁納米粒子。
在一種實施方式中,所述步驟(3)的滾環擴增反應包括連接反應和rca過程;連接反應具體是:加入滾環擴增模板,孵育一段時間使模板兩端與信號探針中的引物序列互補配對,然后加入t4dna連接酶連接反應一段時間使模板兩端連接成環,再使酶失活;rca過程具體是:加入phi29dna聚合酶進行滾環擴增反應,反應一段時間后,使酶失活。
在一種實施方式中,所述檢測的過程為:取滾環擴增產物、hepes緩沖液和氯化血紅素混勻,室溫孵育1h來形成dna酶。在96孔板中加入h2o2母液、魯米諾母液、dna酶和反應緩沖液,混和均勻,用多功能酶標儀檢測相對發光值,根據相對發光值變化對金黃色葡萄球菌進行定量檢測。
在一種實施方式中,所述磁納米粒子的濃度為0.8~1.2mg/ml。
在一種實施方式中,所述捕獲探針的的濃度為80~160nmol/l。
在一種實施方式中,所述連接反應的時間為40~60min。
在一種實施方式中,所述rca過程中滾環擴增反應的時間為40~60min。
在一種實施方式中,所述方法的具體步驟為:
(1)制備捕獲探針功能化的磁納米粒子
將氨基化磁納米粒子加入到戊二醛和pbs溶液中混勻,避光反應,再加入親和素溶液避光反應,獲得親和素修飾的磁納米粒子;再取合成的親和素修飾的磁納米粒子和捕獲探針混勻,室溫條件下孵育、清洗,獲得捕獲探針功能化的磁納米粒子;
(2)目標dna序列與探針生成夾心結構
取捕獲探針功能化的磁納米粒子,加入含目標dna序列的待測液,室溫后孵,再加入信號探針,室溫孵育生成夾心結構,清洗,加水補至10μl;
(3)滾環擴增反應
在步驟(2)制備的混合液中加入12.5μl滾環擴增模板,混勻后孵育,使模板兩端與信號探針中的引物序列互補配對,然后加入t4dna連接酶緩沖液和適量的t4dna連接酶,反應一段時間,使模板兩端連接成環,再于一定條件下孵育使酶失活;清洗后,加入phi29dna聚合酶緩沖液、bsa、dntps、適量phi29dna聚合酶和超純水混勻后,一定條件下進行滾環擴增反應一段時間,并于一定條件孵育使phi29dna聚合酶失活;
(4)滾環擴增產物的化學發光檢測
取擴增產物、hepes緩沖液、氯化血紅素母液和超純水混勻,室溫下孵育形成dna酶。在黑色96孔板中加入h2o2母液、魯米諾母液、dna酶和反應緩沖液,混和均勻,用多功能酶標儀檢測化學發光反應的相對發光值,根據相對發光值變化對金葡菌進行定量檢測。
本發明的優點和效果:
(1)本發明利用夾心雜交原理和磁分離作用,探針修飾的磁納米粒子特異性識別和分離金黃色葡萄球菌的16srdna序列,提高了檢測的準確性和特異性。
(2)本發明結合滾環擴增信號放大作用和dna酶的催化作用,雙重放大,提高了檢測方法的靈敏度。
(3)對金黃色葡萄球菌的檢測限較低,達到4.2×101cfu/ml。
附圖說明
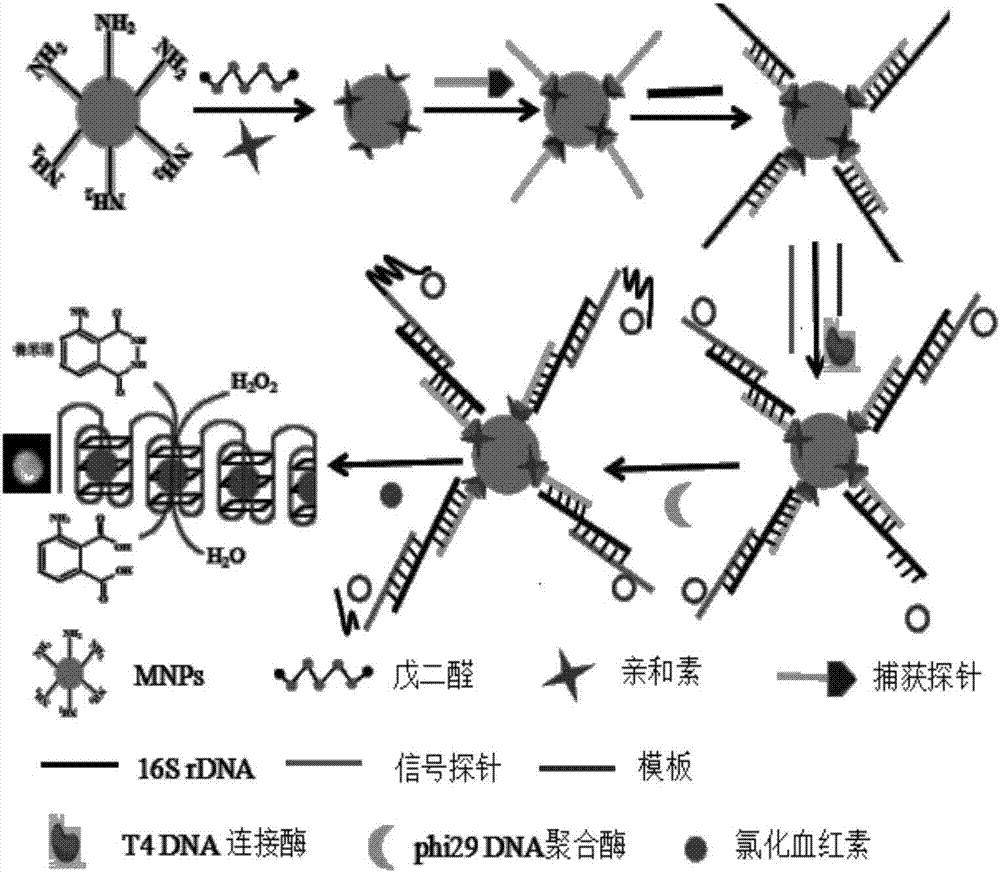
圖1為本發明的金黃色葡萄球菌檢測原理示意圖。
圖2為sybrgreeni熒光驗證dna序列雜交夾心原理圖。
圖3為不同濃度金黃色葡萄球菌與相對發光值的關系曲線。
圖4為tdna檢測的特異性分析。
圖5為金黃色葡萄球菌檢測的特異性分析。
具體實施方案
如圖1所示,為本發明的金黃色葡萄球菌檢測原理示意圖。
利用磁納米分離技術、滾環擴增技術與dna酶技術,構建了基于磁分離滾環擴增合成dna酶檢測金葡菌。
本發明方法:設計兩條不同的dna探針和目標dna(即tdna,如seqidno:1所示)可生成夾心結構,其中probe1為捕獲探針(序列為:biotin-ttttttgaaggctctatctct,如seqidno:2所示),可經生物素-親和素的特異性作用修飾到磁納米粒子上來進行分離;而probe2為信號探針(序列為:agagttgtcaaaggttttttaggatcgtgtggtt,如seqidno:3所示),包含有引發rca的引物序列可與設計的線性rca模板(序列如seqidno:4所示)互補配對,然后開始rca反應,產生與環狀模板互補的長單鏈擴增產物。擴增產物結合hemin形成dna酶(序列如seqidno:5所示),可催化h2o2-魯米諾反應系統發生化學發光反應。
如圖2所示,為本發明方法原理的驗證。捕獲探針功能化的磁納米粒子probe1-mnps可用于捕獲tdna,然后再連接包含rca引物的probe2。在tdna的存在下,probe1和tdna間互補配對形成雙鏈結構,可以使熒光染料sybrgreeni嵌入,并通過熒光分光光度計檢測到較強的熒光信號。相比之下,若沒有加入tdna則反應體系中無雙鏈結構形成,故加入熒光染料sybrgreeni后只能檢測到微弱的熒光信號。當再加入probe2進一步孵育后,形成probe1/tdna/probe2雙鏈夾心結構,則加入熒光染料sybrgreeni后可檢測到更強的熒光信號,表明probe2成功連接到磁納米粒子上。此外,若加入完全錯配的mdna4,則也沒有觀察到顯著的熒光信號,證明只有tdna存在下才能特異性形成夾心結構。
此外,加入10nmol/ltdna的實驗組與未加tdna的空白組經rca反應后經多功能酶標儀m5所檢測到的相對發光值,可以清楚看出,實驗組的相對發光值遠大于空白組,表明只有在tdna存在下,mnps才能修飾上雙鏈夾心結構,并且probe2才能進行后續的rca反應,使得實驗組生成大量的dna酶來催化h2o2-魯米諾反應系統產生強烈的化學發光反應。而當不存在tdna情況下,probe2不能修飾到mnps上,無法進行后續的rca反應和dna酶生成,h2o2-魯米諾反應系統在mnps的催化作用下進行微弱的化學發光反應,只能形成微弱的相對發光值。將實驗組相對發光值減去空白組得到實際相對發光值,且后續實驗中發現實際相對發光值和tdna濃度呈一定線性關系。由此進一步表明,建立的雙鏈夾心結構結合mnps分離技術的特異性強,并利用rca技術和dna酶技術來放大信號,可以用于金葡菌的快速檢測。
下面是對本發明進行具體描述。
實施例1
(1)制備捕獲探針功能化的磁納米粒子
500μl氨基化磁納米粒子加入到150μ戊二醛和1350μlpbs溶液中混勻,避光反應2h,用pbs緩沖液清洗3次,再加入500μl一定濃度的親和素震蕩避光反應6h,pbs緩沖液清洗3次,獲得親和素修飾的磁納米粒子。再取240μl合成的親和素修飾的磁納米粒子和10μl捕獲探針混勻,室溫條件下孵育30min,用te緩沖液清洗3次后,獲得捕獲探針功能化的磁納米粒子。
(2)16srdna序列與探針生成夾心結構
取30μl捕獲探針功能化的磁納米粒子,加入含16srdna序列(即含目標dna)的待測液,室溫后孵育30min,用te緩沖液清洗3次,再加入3μl信號探針,室溫孵育30min生成夾心結構,用te緩沖液清洗3次后,加超純水補至10μl。
(3)滾環擴增反應
在上述制備的混合液中加入12.5μl滾環擴增模板,混勻后在37℃條件下孵育30min,使模板兩端與信號探針中的引物序列互補配對,然后加入2.5μl10×t4dna連接酶緩沖液和適量的t4dna連接酶,37℃反應1h,使模板兩端連接成環,再于65℃條件下孵育10min使酶失活。用te緩沖液清洗3次,加入5μl10×phi29dna聚合酶緩沖液、2μlbsa、2μldntps、適量phi29dna聚合酶和20.5μl超純水混勻后,30℃條件下進行滾環擴增反應1h,并于65℃孵育10min使聚合酶失活。
(4)滾環擴增產物的化學發光檢測
取50μl擴增產物、100μlhepes緩沖液、0.2μl氯化血紅素母液和50μl超純水混勻,室溫下孵育1h形成dna酶,再將dna酶稀釋102倍。在黑色96孔板中加入10μlh2o2母液、10μl魯米諾母液、10μldna酶和170μl反應緩沖液,混和均勻,用多功能酶標儀檢測化學發光反應的相對發光值,根據相對發光值變化對金黃色葡萄球菌進行定量檢測。
(5)對金黃色葡萄球菌檢測的反應條件優化。優化結果為:磁納米粒子濃度為1.0mg/ml,捕獲探針濃度為100nmol/l,擴增時間為50min,連接時間為50min。
(6)金黃色葡萄球菌標準檢測曲線的建立。取500μl不同濃度的金黃色葡萄球菌菌懸液用煮沸法提取dna,再采用本方法進行定量檢測。實驗結果為:在最優條件下,金黃色葡萄球菌濃度在5.5×101~5.5×106cfu/ml范圍內與相對發光值呈線性正相關(如圖3所示),r2=0.991,檢測限為4.2×101cfu/ml。
(7)人工污染實際樣品中金黃色葡萄球菌的檢測:將自來水、桶裝水和牛奶樣品經過紫外照射殺菌后,把不同濃度的金黃色葡萄球菌菌懸液加入其中并混勻。取500μl各樣品用水煮法提取金黃色葡萄球菌的dna,再采用本方法進行化學發光檢測,從標準曲線中求得對應的實際樣品中可被檢測到的金葡菌菌落數,同時將平板計數法作為對照試驗進行比較,檢測結果參見表1。從表1可以得出兩種方法所得的測定結果基本一致,表明構建的檢測金葡菌的新方法可以應用于實際樣品的檢測分析。
表1人工污染實際樣品中金黃色葡萄球菌的檢測結果
實施例2:目標dna(tdna)的特異性分析
特異性是基于夾心雜交原理建立的,即探針與目標序列之間特異性的互補配對作用。
在磁納米粒子濃度為1.0mg/ml,捕獲探針濃度為100nmol/l,擴增時間為50min,連接時間為50mi的條件下,將等體積等濃度tdna(序列如seqidno:1所示)和堿基錯配dna序列(mdna1、mdna2、mdna3和mdna4,與tdna相比,分別為一個堿基錯配dna、兩個堿基錯配dna、三個堿基錯配dna、隨機序列dna;序列分別如seqidno:6~seqidno:9所示,)作為檢測對象,用于評價本實驗的特異性。
如圖4所示,不同dna序列檢測得到不同的相對發光值,從中可看出tdna對應的相對發光值明顯高于mdna1、mdna2、mdna3和mdna4。此外,根據圖4還可以推斷出,堿基錯配的數目越多,檢測得到的相對發光值越低,其中隨機序列mdna4與空白對照結果相近。以上分析結果表明,堿基的錯配會影響探針與tdna的互補配對作用,從而影響probe1-mnps捕獲tdna以及tdna與probe2的雜交作用,以及rca反應的進行和dna酶的生成,最終使得相對發光值的降低,從而可確保對tdna檢測的特異性。故推斷,基于序列如seqidno:1所示的tdna構建的檢測方法具有較高的特異性。
實施例3:金黃色葡萄球菌的特異性分析
基于核酸探針與金葡菌16srdna特異性互補配對的原理。
使用金黃色葡萄球菌、沙門氏菌、大腸桿菌、蠟樣芽孢桿菌和痢疾志賀氏菌作為待測樣品,采用實施例1的方法對檢測效果進行了考察和比較,并以te緩沖液為空白對照。
由圖5可以看出金葡菌的相對發光值遠遠高于沙門氏菌、大腸桿菌、蠟樣芽孢桿菌和痢疾志賀氏菌,表明實施例1的方法對金葡菌具有良好的特異性。
雖然本發明已以較佳實施例公開如上,但其并非用以限定本發明,任何熟悉此技術的人,在不脫離本發明的精神和范圍內,都可做各種的改動與修飾,因此本發明的保護范圍應該以權利要求書所界定的為準。
序列表
<110>江南大學
<120>一種磁分離rca合成dna酶檢測金黃色葡萄球菌的方法
<160>9
<170>patentinversion3.3
<210>1
<211>29
<212>dna
<213>人工序列
<400>1
cctttgacaactctagagatagagccttc29
<210>2
<211>21
<212>dna
<213>人工序列
<400>2
ttttttgaaggctctatctct21
<210>3
<211>34
<212>dna
<213>人工序列
<400>3
agagttgtcaaaggttttttaggatcgtgtggtt34
<210>4
<211>66
<212>dna
<213>人工序列
<400>4
accgactcaaaacccaacccgccctacccaaaacccaacccgccctacccaaacatacac60
acagca66
<210>5
<211>30
<212>dna
<213>人工序列
<400>5
tggctgagttttgggttgggcgggttgggt30
<210>6
<211>29
<212>dna
<213>人工序列
<400>6
cctttgacaactttagagatagagccttc29
<210>7
<211>29
<212>dna
<213>人工序列
<400>7
cctttgacaactttagagatagatccttc29
<210>8
<211>29
<212>dna
<213>人工序列
<400>8
cctttaacaactttagagatagatccttc29
<210>9
<211>29
<212>dna
<213>人工序列
<400>9
ctactaatgctcctggattcatcacttca29
- 還沒有人留言評論。精彩留言會獲得點贊!